

2013年,单细胞测序技术开始成为科研界主流关注的焦点。
前言
2013年,单细胞测序技术(single-cell sequencing)荣膺《自然-方法》年度技术。单细胞测序技术有助于我们剖析细胞的异质性。它可以揭示肿瘤细胞基因组中发生的突变及结构性变异,而这些突变和变异往往有着极高的突变率。有了这些信息,我们就可以描述肿瘤细胞的克隆结构,并追踪疾病的进展及扩散范围。本文将介绍2013年单细胞测序技术在人类早期发育、癌症以及神经科学研究等几个重点领域的最新应用成果。
1. 单细胞测序技术简介
本节将概述如何获得一个单细胞的基因组及转录组。
单细胞基因组及转录组测序所需要的测序样本量要比单细胞中本身所含有的基因组及转录组分子高出好多个数量级,所以这对核酸扩增技术(amplification technology)也是一大考验。面对如此微量的分子,任何降解、样品损失、或者污染都会对测序质量带来非常严重的影响。而且多重扩增又容易带来试验误差,比如基因组或转录组覆盖不均一、背景噪声以及定量不准确等问题。
最近所取得的技术进步有望部分解决上述问题,使单细胞测序技术能够走进更多的实验室,解决更多领域的科学问题。比较罕见的细胞、异质性的样本、与遗传嵌合或突变相关的表型、不能人工培养的微生物,这些都是单细胞测序技术能够一展所长的研究平台。使用单细胞测序技术能够发现克隆突变(clonal mutation)、隐藏的细胞类型,或者在大块组织样品研究工作中被“稀释”或平均掉的转录特征。
1.1 选择恰当的细胞
说到分离单细胞,显微操作(micromanipulation)无疑是一项非常精确的技术,而且利用毛细管(microcapillary)可以直接吸取细胞内容物,但是这项操作也需要耗费大量人力。很多组织解离之后都能够制成单细胞悬液,这种单细胞悬液很容易操作,而且可以用细胞分选器(cellsorter),根据细胞表面表达的特异性分子标志物对细胞进行分类富集操作。这种策略也被用来分离非常微量的循环肿瘤细胞。
1.2 单细胞转录组策略
现在有很多单细胞RNA测序操作流程可供选择,不过不管采用何种策略,首先都需要通过逆转录反应,利用RNA合成出cDNA。然后才会有所区别,比如有一些方法是对整个转录子进行测序,有一些方法只针对转录子的5'和3'端进行测序。不论采用何种方法,目的都只有一个,那就是捕获原始的RNA分子,然后均一的、准确地对其进行扩增。核酸的捕获效率主要受到逆转录反应的影响,不过我们可以使用更小的反应体系,选择更好的逆转录酶来进行改善。另外,采用模板转换技术(template switching)也能够保证被捕获的绝大部分转录子都是全长片段。减少反应循环数也能够改善核酸扩增反应,还可以借助“抑制PCR(suppression PCR)”技术减少引物扩增,或者将取自不同样品的cDNA(这些cDNA都是分别做好标记的)混合到一起,提高起始反应模板浓度,用体外转录技术进行线性扩增(linear amplification)。另外,还可以利用特有的分子识别序列(molecular identifier sequences)对每一个RNA分子进行标记,这样即便在经历了非均一的扩增之后,我们还是能够对原始的RNA分子数量进行绝对定量。
1.3 单细胞基因组策略
全基因组扩增(whol e-genome amplification)的起始反应产物更少,只有一个DNA分子。这样在扩增反应时就难免出现不均一的问题,即可能在基因组中某些位点会扩增多次,而另外一些位点则无法扩增。解决这个问题最常用的办法就是多重置换扩增技术(multiple displacement amplification, MDA),即使用随机引物,让这些引物与基因组广泛结合,同时使用一种特定的聚合酶,这种聚合酶能够置换与它自身附着在同一模板上的DNA链片段,形成一种反复分支结构(iterative branching structure),扩增出大段的DNA。早期循环对整个扩增反应的均一性起到了决定性作用。有一种扩增技术采用了一种独特的引物,这样能够生成闭合环状的扩增子(amplicon),而且这种扩增产物不会再进一步复制,等于是在进行PCR扩增反应之前先进行几轮线性扩增反应。将反应按比例扩大,同时对反应情况进行实时监控都有助于改善基因组扩增成功率低的问题,另外减少扩增次数,准备更少模板的测序文库也是一个比较值得发展的方向。
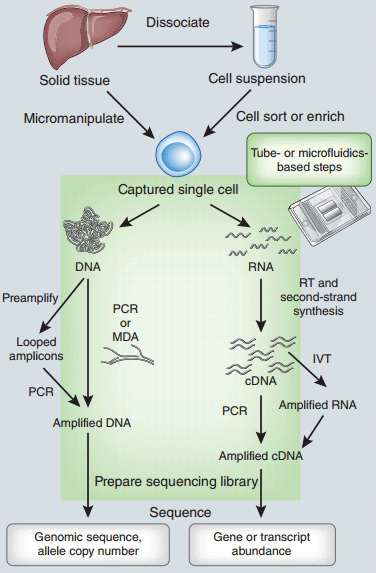
单细胞基因组或转录组扩增及测序工作流程图。
1.4 一个细胞解决所有问题
在单细胞研究工作中,扩大试验规模是确保采集足够多的生物多样性信息的关键。微流体设备(microfluidics)或微孔板技术(microwell technology)能够提供标准化、高通量的选择,而且由于这种设备的反应体积通常都比较小,所以反应效率也都比较高。不过微流体设备也有一定的限制,只能处理某些特定大小的细胞样品。当然,将待测分子用生物条码(barcoding)标记之后混合起来进行分析也是一条处理通量的途径。单细胞核酸扩增及测序技术正在不断成熟、完善之中。我们相信,随着单细胞试验操作变得越来越容易,成本变得越来越低,会有更多的科研人员选择使用单细胞测序技术,这将会像PCR技术一样成为每个实验室里的常规试验操作,帮助我们以更高的分辨率去研究问题、解决问题。
2. 单细胞测序技术——科研界主流关注的焦点技术
单细胞基因组测序技术及单细胞转录组测序技术又掀起了一波新的科研浪潮,让科研人员们能够以新的视角看待发育、肿瘤及神经科学问题。
对于不孕不育症夫妇而言,孕育一个孩子是非常困难的事情,而且这也会让他们的情感饱受折磨。即便他们怀孕了,也不是高枕无忧的,因为这些家长需要担心另外一个问题,如何生育一个健康的宝宝。对于那些存在遗传风险,需要借助体外受精技术(in vitro fertilization, IVF)辅助的父母而言,胚胎植入前遗传学诊断(preimplantation genetic diagnosis, PGD)技术(即从早期胚胎中取出一个细胞进行遗传学疾病筛查的技术)是孕育出健康下一代的保证,虽然目前PGD技术也只能够对基因组中的一个、或少数几个位点进行筛查和诊断。由于取自早期胚胎的细胞数量都不会太多,所以极其珍贵,临床医生们必须从这些宝贵的细胞中尽可能地获取有价值的信息。
而单细胞全基因组测序技术(single-cell whole genome sequencing method)就有望解决早期胚胎发育及其他科研领域里存在的这些重要的问题。由于单细胞分离技术以及单细胞中痕量的DNA或RNA扩增及测序技术的进步,科研人员们得以对单细胞的整个基因组或转录组(而不是少数几个位点)以前所未有的高分辨率进行扫描和研究。
美国哈佛大学(Harvard University)的Sunney Xie等人就是在IVF工作中进行单细胞基因组测序研究的课题组之一,他们用第一极体和第二极体(所谓极体指的是受精卵分裂时“遗弃”的细胞成分,可以反映染色体的健康状况)验证了他们新开发的全基因组扩增技术。Xie等人最近发表的文章介绍了他们对8位女性供体的研究成果,研究发现极体活检(polar-body biopsy)和单细胞测序都能够准确地反映胚胎染色体非整倍体(aneuploidy)的情况,其中就包括唐氏综合症(Down's syndrome)这种染色体数目过多的情况,也包括染色体丢失,或者遗传自父母的单核酸突变(single-nucleotide variation)等情况(Cell, doi:10.1016/j.cell.2013.11.040 19 December 2013)。Xie发现染色体非整倍体平均只需要在每一百个基因组位点中挑出一个位点进行测序就足够了,所以这要比传统的方法成本更低,而且准确性也会更高。
Xie与这篇论文的合作者——中国北京大学(Peking University)的Fuchou Tang和中国北京大学附属第三医院(Peking University Third Hospital)的Jie Qiao针对这些接受IVF帮助的女性开展了一项临床研究。他们对这些志愿者的胚胎极体进行了基因组扩增和全基因组测序研究,以此来判断胚胎是否健康,是否适于进行移植、受孕。据Xie介绍,如果将时间提前两年,在临床上开展这项研究工作几乎是不可能的,当时有大批没有遗传问题,可是不能受孕的夫妻给他发邮件寻求帮助。目前这次临床研究的第一位婴儿将在年内降生。Xie指出,他并没有想到他们的技术能够这么快地走向临床,给患者们提供切实的帮助。”

Sunney Xie很快就能够看到他们课题组开发的基因组扩增技术被应用到临床PGD实际工作当中的表现情况了。
2.1 2013年测序技术回顾
单细胞测序技术可谓是科技发展史上的一大创举。一个细胞里的DNA或RNA仅仅处在皮克(picograms)级的水平,这么少的量远远达不到现有测序仪的最低上样需求。因此科学家们必须先对单细胞内的微量核酸分子进行扩增,而且必须保证尽可能少地出现技术误差,以便开展后续的测序及其他研究。直到最近,也还是只有少数几位专家相信能够对单细胞进行可靠的研究。
虽然早在几年前就开始有研究团体在宣传、推广单细胞基因组及转录组测序技术,但是这些技术是最近这两年才开始被大范围接受,其中就包括从事神经科学研究、肿瘤及微生物生态学研究的科研人员。据美国Fluidigm公司的联合创始人,斯坦福大学(Stanford University)的Stephen Quake介绍,几乎从PCR技术诞生的第一天开始,就不断有人尝试用PCR技术进行单细胞基因表达研究及单细胞基因组研究。但是由于种种原因,单细胞测序技术直到现在才算是刚刚起步。
DNA和RNA扩增技术的不断改进,尤其是最近这两年新出现的进步给刚刚涉足这个领域的科研人员在开展试验时提供了非常丰富的选择。工业界也提供了无数种商业化的、而且价格低廉的单细胞核酸扩增试剂盒及读取技术。Fluidigm公司就在2013年推出了世界上第一款单细胞RNA测序自动化准备系统(single-cell automated prep system for RNA-seq)。所有这些技术上的进步极大地降低了科研人员们进入单细胞研究这个领域的技术门槛。瑞典卡罗林斯卡研究院(Karolinska Institutet in Sweden)的Rickard Sandberg在谈到单细胞RNA测序时说道:“大家等这一天已经等了好几十年了。直到今天,由于技术的进步,这些试验才变得足够简单,而且成本也能够让大家接受,所以才能够走进千千万万个实验室。”
进行单细胞研究的核心问题其实是:为什么要进行单细胞研究?这主要是因为如果将成千上万个细胞混在一起进行研究,就会模糊我们对大脑、血液系统、免疫系统,及其组成这些系统的细胞之间异质性(heterogeneity)的认识。美国宾夕法尼亚大学(University of Pennsylvania)的James Eberwine就认为,当你的研究深入到单细胞层面时,你就会失去对整个系统的把控,但是如果你能够从整个系统中挑选多个不同的单细胞进行研究,则可以重建出整个系统,而且这种重建过程能够提供更多更有价值的信息。有大量很难对大块组织进行研究的科研领域也都会从这些最新的单细胞研究技术中获益。这种单细胞测序技术不仅有助于我们认识细胞之间的差异,还可以为我们提供一个新的比较层面,这也是大家期盼已久的,能够重新定义细胞类型的层面。
可与大家这种极高热情相伴的却是各种各样的技术难题,包括单细胞分离、基因组或转录组扩增,以及数据解读等各个方面。试验成本也是需要考虑的一大因素。通常来说,对细胞进行分析时所需要的细胞数量要比对组织进行分析时所需要的组织数量多很多,所以在决定是否应该进行单细胞研究时一定要谨慎,要根据实际情况做出合适的判断。“我们真的需要进行单细胞研究吗?如果答案是否定的,那就不要进行单细胞研究。单细胞研究非常贵,而且你会碰到很多的变数。”美国博大研究院(Broad Institute),同时也在麻省理工学院任职的Paul Blainey这样说道。
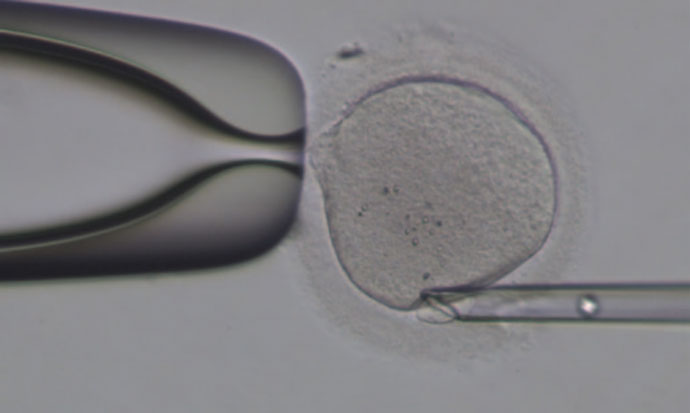
对体外受精卵分裂产生的极体进行基因组测序能够为临床医生们进行胚胎植入前的遗传学诊断和筛查提供非常有价值的帮助。
2.2 从少数几个RNA分子开始
对细胞的转录组进行测序,关键在于能否利用细胞内的RNA扩增出大量的cDNA。然而,捕获少量的RNA分子制备cDNA,以及大量扩增这些cDNA分子的工作很难做到平等和高效。
1990年,Norman Iscove的课题组首次证实对单细胞进行转录组分析是可行的,他们用 PCR技术实现了对cDNA分子的指数级扩增。在20世纪90年代初期,Eberwine等人发明了一种新技术,能够从单个的活神经元细胞中获得cDNA,并且再以这些cDNA为模板转录生成RNA,实现RNA的线性扩增。随着芯片时代的来临,科学家们用这些线性、和指数级扩增技术对单细胞之间的基因表达差异进行了大量的比较和研究。
2008年时出现了高通量RNA测序技术,不久之后,科研人员们就将这种技术与前面发展起来的核酸扩增技术结合起来,对单细胞转录组进行了更加精细的研究。2009年,当时在英国剑桥大学Gurdon研究所(Gurdon Institute at the University of Cambridge)M. Azim Surani实验室工作的Tang通过对单个小鼠卵裂细胞(blastomere)的研究发现,与芯片技术相比,利用单细胞转录组技术可以多发现数千个基因的表达情况(Nat. Methods 6, 377–382, 2009)。
就在同一年,美国冷泉港实验室(Cold Spring Harbor Laboratory)也召开了第一次单细胞大会,参加大会的有科研人员、技术开发人员,以及涉足单细胞研究领域的先驱们,参会者一共还不到50人。据现在在美国弗吉尼亚大学(University of Virginia)工作的Mike McConnell回忆,每一个人都试过做 RNA测序,也尝试过对测序结果进行分析,想从中找出有价值的、可重复的结论。
不过技术的发展经历了很长一段时间,现在终于有了一整套单细胞测序的操作流程和各种商业化的试剂盒产品。瑞典卡罗林斯卡研究院的Sten Linnarsson认为,纯粹技术上的发展到今年已经达到了顶点,现在是考虑如何将这些技术应用到实际工作当中。有很多课题组瞄准的可不是几百个细胞,他们想要研究上万个细胞。
比如 Kun Zhang课题组承担的、由美国国立卫生研究院公共资金(US National Institutes of Health Common Fund)资助的单细胞研究项目(Single Cell Analysis Program)就打算对取自人类大脑皮质三个不同区域里的10,000多个细胞进行全转录组分析。Zhang等人计划根据转录子对细胞类型进行分类(可能还会对细胞类型进行重新定义),并且将这些转录子重新定位到不同的大脑组织切片里。当然单细胞RNA测序这项技术本身已经不再是障碍了。据Zhang介绍,如果你有好的细胞,如果你想进行转录组研究,那么你会有很多种选择,帮助你达成目标。不过通常而言,如何在人死后提取神经元细胞,并尽可能减少RNA的降解,保持大脑组织正常的空间结构,这也都是需要解决的问题。 Zhang等人也在从事这方面的工作,正在对几种不同的技术进行比较。
2.3 基因组扩增技术
开发一项新的单细胞全基因组扩增技术是需要一定的时间的,这是因为在一个细胞内,通常都只有一至两个DNA的拷贝,所以直到2005年才出现单细胞全基因组扩增技术,这要远远落后于单细胞RNA扩增技术。当时Roger Lasken的团队成为世界上第一个成功完成单细胞DNA扩增及测序的团队,他们当时使用的是自己开发的多重置换扩增技术(multiple displacement amplification, MDA)对大肠杆菌进行试验。这项工作给微生物学家带来了极大的激励,他们利用这项技术对各种不能人工培养的微生物进行了测序研究,获得了大量的参考基因组序列(reference genome)。
MDA作为最常用的技术和策略一直沿用至今,该技术用到了Phi29等聚合酶,能够使经退火、结合到基因组上的任意随机引物不断延伸。每一种聚合酶都能够置换临近的延伸链,形成大量的、覆盖多个小片段的、长达7至10kb的大片段产物,用来进行DNA测序。
到了2011年,科研人员们将单细胞基因组扩增技术与高通量测序技术结合起来开展研究。Nicholas Navin当时就在美国冷泉港实验室的Michael Wigler课题组工作,他在取自两位乳腺癌患者的乳腺癌肿瘤细胞中发现了大段的基因组DNA缺失或重复突变,即拷贝数变异(copy-number variant, CNV),该研究的分辨率达到了50kb(Nature 472, 90–94,2011)。
在单细胞基因组测序工作中最大的困难就是某些DNA片段的扩增效率要远远高过另外一些DNA片段。Xie等人在2012年又发明了一种新的多重退火环状扩增循环技术(multiple annealing and looping-based amplification cycles, MALBAC),该技术首先需要进行5轮MDA预扩增,然后就可以使新获得的扩增产物形成闭合的环状分子(Science 338,1622–1626, 2012)。由于这些环状分子不能够被进一步扩增,所以整个扩增过程就变成了线性扩增。然后再进行常规的PCR扩增,由于此时采用的模板更加均一,所以在进行PCR扩增时就不易造成较大的差异。Xie等人用这种MALBAC技术获得的人类基因组扩增产物能够达到93%的覆盖度,同时也在单个肿瘤细胞中检出了CNV突变。
很快,科学家们就能够对单细胞的基因组进行更加深入的研究了,他们将能够发现更小的缺失和重复突变,乃至单碱基突变。虽然基因组均一扩增还是一个问题,但是专家们相信,缩小反应体积应该可以带来一定的帮助。
比如美国加州大学圣地亚哥分校(University of California, San Diego)的Zhang等人最近就介绍了一种MIDAS技术,即微孔板置换扩增系统(micro-well displacement amplification system),使用这套系统可以用纳升级的反应体系同时进行数千个MDA反应(Nat. Biotechnol., 31,1126–1132, 2013)。科研人员们可以手工、或者用机械手取出这些扩增产物,进行测序。借助这套MIDAS系统,Zhang等人课题组只进行了很少的测序工作就在人类神经元细胞中发现了单拷贝变异(single-copy-number change),分辨率达到了1至2MB。

这套MIDAS系统是一种高通量的单细胞分离、扩增及测序技术。
2.4 细胞表达差异
在美国博大研究院(Broad Institute),Aviv Regev与Joshua Levin等人在开始单细胞RNA测序工作之前,先利用质量很差、降解严重的组织样本对多种RNA测序技术进行了比较,最后她们决定采用Smart-Seq技术对骨髓来源的树突状细胞(dendritic cell)进行研究。这些树突状细胞是一种有丝分裂后的免疫细胞,能够对抗原产生非常强烈的转录反应。
Regev等人一共选择了18个细胞,耗时一周分批进行了试验。她们之前尝试了各种方法,最终都失败了。可是这一次却一次就成功了。研究发现,每一个细胞都会统一表达所谓的持家基因(‘housekeeping’genes),但是每一个细胞也都有各自独特的表达谱,与免疫调控功能相关的基因在有些细胞里的表达水平非常高,可是在有些细胞里却压根不表达。之前还从来没有在树突状细胞中发现这种两极分化的现象,因为一直以来都是对一堆细胞进行研究,细胞之间的差异全部被平均掉了。该研究成果于去年6月得以发表,该文章首次报道了一种“隐藏的”细胞类型,即非常罕见的“第一应答者细胞(first responder)”(Nature 498, 236-240, 2013)。从更广义的角度来说,这一发现有助于我们重新认识这些树突状细胞,以及它们的信号通路和功能。

单细胞RNA测序技术第一次尝试就取得了成功。
——美国博大研究所Aviv Regev
单细胞转录组测序也能够帮助科研人员研究发育早期的基因表达与调控情况,而且借助这项技术还能够以前所未有的精细程度对罕见的样品开展科学研究。比如美国加州大学洛杉矶分校(University of California, Los Angeles)的Guoping Fan与他在中国的合作者们在去年8月发表的一篇文章就对33个单细胞进行了转录组测序研究。这33个细胞全都取自处于发育不同阶段的胚胎,他们根据测序结果确定了发育初期基因的表达顺序,还发现了人类与小鼠胚胎发育过程中基因表达时限上的差异(Nature 500, 593–597, 2013)。

单细胞测序技术是一项非常强大的技术,可以帮助我们发现肿瘤细胞里的基因组变异。
——美国德克萨斯大学MD Anderson癌症中心Nicholas Navin
与此同时,Tang的课题组也在从好几个人类早期胚胎中仔细地分离细胞标本,并且对这些细胞挨个进行单细胞转录组测序。据Tang介绍,他们非常紧张,因为这些标本全都来之不易,非常珍贵。不过他们的工作也获得了回报,他们发现了2700多个新的长非编码RNA(long noncoding RNA)分子,这些分子可能都与早期基因调控作用有关(Nat. Struct. Mol. Biol. 20, 1131–1139, 2013)。据Tang介绍,在此之前,所有的单细胞RNA测序工作还都只是针对已知基因进行分析,充其量也仅仅增加了已知基因的可变剪接亚型(alternative splicing isoforms)而已。
2.5 混合的肿瘤细胞
从疾病预后判断到病情监测,肿瘤研究人员都能够从单细胞测序技术那里获得巨大的帮助。我们都知道,肿瘤细胞的突变速率非常快,而且肿瘤组织是一种高度异质性的组织。确定肿瘤组织中存在哪些细胞亚群(或者叫克隆)具备转移能力,哪些克隆对化疗药物是敏感的,这些信息对于临床工作都非常有帮助。尤其针对隐藏在人体循环系统里的循环肿瘤细胞(circulating tumor cell, CTC)进行全基因组或者转录组测序最有帮助,因为这些CTC细胞就是导致肿瘤转移的元凶,有关它们的信息对于疾病的诊断、监测和治疗都至关重要。
比如Navin于2011年在《自然》(Nature)杂志就发表过一篇文章,介绍了他们的单细胞基因组研究成果。他们发现CNV突变与肿瘤的进化模式有关,肿瘤在稳定增长之后会突然发生基因组失稳。据现在在美国得克萨斯大学MD Anderson癌症中心工作的Navin介绍,这一发现让他们非常吃惊,因为他们一直认为肿瘤细胞一直在缓慢地积累突变。这次研究工作也证实,单细胞技术非常强大,至少能够帮助他们发现人体单个肿瘤细胞里的基因拷贝数变异。Navin与他的合作者们还在继续对三阴型乳腺癌患者进行研究,主要想了解CNV方面的情况,同时也希望能够更好地了解肿瘤转移的问题。
除了Navin等人之外,还有其他几个课题组也都在利用单细胞测序技术开展与肿瘤相关的研究工作。比如前面介绍过的Xie就与中国北京大学的Fan Bai,以及美国哈佛大学的Jie Wang一起,在一种肺癌亚型(不包括其它亚型)的CTC细胞中发现了一种特定的CNV突变(Proc. Natl. Acad. Sci. USA, doi:10.1073/pnas.1320659110, 9 December 2013)。Xie认为,这些最新的进展都有助于我们开发早期诊断产品和技术。

Mike McConnell在单个人脑神经元细胞中发现了大段的DNA缺失或重复突变。
转录组上的差异也有助于我们认识肿瘤的进展情况。比如Sandberg的团队就使用他们自己开发的Smart-Seq技术对单个CTC细胞进行了RNA测序研究,并对他们的这套方法进行了验证。使用最新版的Smart-Seq2技术,他们能够以比以前更低的成本观察更多的细胞。由于观测的细胞数更多,所以让从事CTC研究的科研工作者们头痛不已的试验误差问题也能够得到更好的控制。据Sandberg介绍,他们真的希望拿出一套更加系统的解决方案,帮助大家更好地认识CTC细胞的异质性问题,帮助大家更好地认识CTC细胞进入血液循环系统时的基因表达情况。

Wolf Reik希望表观遗传学技术也能够早日达到单细胞检测水平。
比基因组和转录组研究更困难的就是以化学标志物形式附着在基因组上,并对基因的表达实施调控的表观基因组(epigenome)研究了。虽然目前的表观遗传学技术还达不到单细胞研究水平(因为传统的表观遗传学研究技术都会使DNA降解),但是科研人员们还是迫切希望看到单个肿瘤细胞的表观基因组情况。Tang的科研团队开发了一种可以对单细胞全基因组内的DNA甲基化修饰情况进行研究的新技术(Genome Res. 23, 2126–2135, 2013)。Tang认为,表观基因组研究真的也需要单细胞技术,只有这样,科研人员们才能够了解这个肿瘤细胞与它周围的肿瘤细胞有什么差别,而且这种差别是因为甲基化修饰引起的,还是因为其它机制引起的。英国Wellcome基金会Sanger研究所(Wellcome Trust Sanger Institute)的Wolf Reik团队对 50至100个细胞的甲基化组(methylome)情况进行了分析,他表示他真的很想再往前走一步。
2.6 大脑中的“禁区”
神经元细胞是最新一个被用来进行单细胞研究的对象,科学家们其实也不太清楚能够通过这些研究获得怎样的信息和结论。也是直到最近才开始有试验证据表明,神经元细胞之间也具有不同的基因组。虽然有这些研究成果,但是科学家们对神经元细胞的这种多样性也还是一头雾水。早在2001年,当时还在美国加州大学圣地亚哥分校(University of California, San Diego)工作的Jerold Chun就在小鼠的大脑中发现了染色体非整倍体现象,随后又于2005年在人类大脑细胞当中发现了同样的现象。据当时在Chun实验室读研究生的McConnell介绍,拿到这些结果之后,他们也没人知道下一步该怎么办。他们等于是发现了冰山的一角,如果细胞里存在非整倍体现象,那么一定会有很多的基因突变,或者基因组突变。
几乎就在同一时间,另外一帮科研人员发现,在人类基因组当中,平均每一个基因组里都含有80~100个具有潜在活力的L1元件(这是一种可以在整个基因组当中自我复制、自我粘贴的DNA元件),而且在大脑神经元细胞当中,这些L1元件都是有活性的。该研究,以及其它一些研究成果都证明,基因组至少是具备多样性可能的,但是这种变异的程度究竟有多大,没人说得清楚。
据美国国立精神卫生研究院( US National Institute of Mental Health)的 Thomas Insel介绍,他们还只是刚刚开始尝试去了解大脑细胞的分子多样性问题。在这个领域单细胞研究技术起到了关键性的作用,不仅仅是在确定神经元细胞和神经胶质细胞的(分类)类型方面,同时也有利于我们了解体验和发育对大脑某个区域里的基因表达有何作用。
科学家们可以用好几种方法发现单细胞基因组变异情况。美国哈佛大学医学院(Harvard Medical School)的Christopher Walsh团队就对300个取自死者大脑的神经元细胞进行了单细胞L1元件插入研究(Cell 151, 483–496, 2012)。他们只发现了几个 L1插入元件,这说明L1元件应该不是导致基因组多样性的主要原因,但至少在大脑皮质细胞和尾状核(caudate nucleus)细胞里是这样。
2013年,另外几个课题组也对单个人类神经元细胞进行了全基因组扫描研究。比如在2013年11月发表的文章就对3名健康人大脑的110个额皮质(frontal cortex)神经元细胞进行了全基因组测序研究,结果相当令人吃惊,他们发现在神经元细胞里有大量的大段CNV突变(Science 342, 632–637, 2013)。对源自健康人皮肤细胞的神经元细胞进行的研究也发现了同样的情况,而且这些神经元细胞里的CNV要比其来源的皮肤细胞更多,这说明这种源自iPS细胞的神经元细胞是一种非常好的研究材料,适合用于开展细胞多样性方面的研究工作。
实际上,虽然有了这些发现,但是神经科学家们还是很头疼,因为他们不知道这些体细胞突变意味着什么。美国弗吉尼亚大学(University of Virginia)的遗传学家Ira Hall也是去年这篇发表于《科学》上的文章的合作者之一,他认为这些研究意味着大脑对影响和干扰的抵抗力很弱,另外,遗传嵌合现象(genomic mosaicism)也能够影响人们罹患肿瘤和其它疾病的风险。为了明确大脑中哪些部位与其它部位相比更容易受到干扰,以及大脑不同区域间的差异有多大,科研人员们还得研究更多的细胞才能够找到答案。现在就在从事这方面研究的McConnell认为现在还是一无所知。
2.7 概念验证之后的工作
虽然单细胞技术已经有可能解决很多生命科学领域的重大问题,但是技术上的进步还远远没有结束。比如科研人员就必须研究如何将真正的生物学差异与试验技术本身的背景噪音区分开。瑞典KTH皇家理工学院(KTH Royal Institute of Technology in Sweden)的Joakim Lundeberg(他们实验室就曾经开发过组织RNA测序技术)就认为,单细胞RNA和DNA测序技术还远远算不上足够强大,他表示,他们还需要在一次试验中对更多的单细胞进行分析,以便解决背景噪声问题,这至少也能够加深他们对同一个组织里不同细胞之间差异的了解。
由于存在方方面面的问题,比如细胞分离、数据运算、以及用于不同领域时出现的特异性问题等等,所以Blainey希望在未来的几年里单细胞研究技术还能够有更大的进步。
对于新进入这个领域的人而言,光是选择哪一种转录组测序技术可能就够他们头疼半天的了。关于这个问题,应该视研究目的而定,比如是想对多个细胞进行分析,找出同型的转录子,还是想发现低丰度的RNA。“不过有多种方法可供选择总归是件好事。”Quake这样说道。在去年10月,Quake的课题组发现,如果将预处理时的反应体积控制在纳升级(他们使用的是Fluidigm公司提供的C1系统),那么单细胞qPCR技术和单细胞RNA测序技术的检测效果是差不多的(Nat. Methods 11,41–46,2014)。“这对于我们整个试验操作的可信度而言是一个重大的好消息。”Quake补充道。
随着商业化产品的推出,以及各个实验室经过多年实践总结出了自己的“独门秘笈”,基因组扩增技术的选择也在同步改善。不过由于每一个人使用的进行基因组扩增的技术都不一样,所以很难对不同的研究成果进行直接的比较。比如Xie就认为,他们感觉MALBAC技术要比MDA技术更好,但是这也要取决于你是如何进行MDA试验的。不过随着技术的不断进步,这两种技术都将会过时被淘汰,但我们也会继续改进这些技术,MALBAC一定会赢得最终的胜利,我们会让这项技术变得更好。
与此同时,从事肿瘤研究、脑神经科学研究、微生物研究、以及从事药物开发和其他领域研究的科研人员也都会从这些技术进步当中受益。而且这些技术进步也会吸引众多优秀的人才加入单细胞研究领域,比如已经在表观遗传学研究领域颇有建树的Reik等。Reik在去年才第一次参加单细胞学术会议,而在此之前还从来没有接触过单细胞研究,看到这么多新技术,Reik感到非常激动。他指出,最开始人们会因技术本身而激动,过不了多久,人们就会利用这些新技术去解决重要的生命科学问题,那将是更加令人激动的事情。
3. 单细胞分析技术——认识遗传多样性的利器
技术上的新进展已经让单细胞基因组测序技术(single-cell genome sequencing)逐渐成为了一项主流的检测手段,该领域的研究工作已经初步揭示出细胞之间在基因组结构(genetic architecture)与遗传变异性(genetic variability)方面的差异,这也反映出基因组并非一成不变的天然本质。
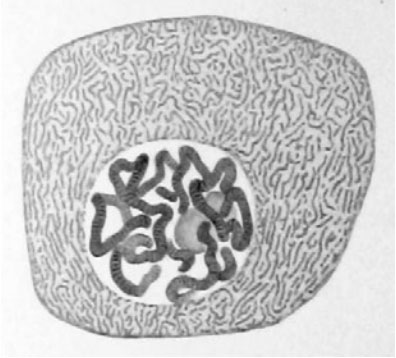
Flemming在1882年时发表的文章中绘制的单细胞基因组染色体模式图。
其实单细胞基因组分析这个项目很早就出现了,早在1882年就有人报告了昆虫唾液腺的单细胞图像,该图展示了多线染色体(polytene chromosomes)的带状结构。到了1935年,Calvin Bridges又发表了一幅类似的果蝇(Drosophila)细胞基因组图片,从这幅图中可以看出个体之间、品系之间,以及种系之间都存在大范围的基因组重排(genomic rearrangements)现象。最近研究人员也开展了大量的单细胞研究工作,使用的主要技术手段包括PCR和其它生化扩增技术。其中比较知名的工作包括在20年前开展的对单个精子细胞(single sperm cell)进行的重组热点(recombination hot-spot)研究,以及现在在人工辅助生殖工作中常规开展的胚胎植入前的胚胎单细胞遗传诊断工作(preimplantationgeneticdiagnoses)。既然单细胞检测技术已经发展了一个多世纪了,为什么现在才突然火起来呢?
我们认为这应该与最近取得突破的单细胞基因组测序工作有关。这主要包括以下三个方面:技术进步使全基因组及转录组扩增的效率大幅度提高;DNA测序技术的跨越式发展使得测序的通量更高,测序的成本更低;微流体技术(microfluidics)和荧光活化细胞分选技术(fluorescence-activated cell sorting)等不断涌现的新型单细胞试验技术。最近这5年,全世界各个实验室里出现了一大批单细胞研究论文,包括单细胞基因表达研究、单细胞基因组分析研究,以及商业化的服务等,这些工作对新技术的推广起到了非常重要的作用。单细胞基因组分析现在就是一个非常有影响力的技术,而且涉及了很多的方面,比如微生物生态学(microbial ecology)、肿瘤、产前诊断以及人类基因组结构及变异等。接下来我们将重点介绍这几个方面的最新进展,以及未来可能的发展方向。
3.1 单细胞生物的单细胞测序
微生物生态学是最适合进行单细胞基因组测序的研究项目,因为据估计,绝大多数(99%的物种)微生物都是无法进行人工培养的。这些不能培养的微生物被科学家们形象地称作生物界的“暗物质(dark matter)”,因为我们只能根据对标志基因(marker-gene)序列的检测来间接地“观察”这些暗物质。虽然元基因组技术(metagenomic approaches)有助于我们了解这种复杂环境里的基因组成情况,但是物种与基因之间的关系还是不得而知,因此只有借助单细胞基因组技术才能够了解单细胞生物(unicellular organism)与自身基因组功能之间的关系。这也说明我们现有的基因组数据库还相当欠缺,有大量的遗传与进化多样性信息都没有被收入在内。
科学家们开展的第一个不能人工培养的单细胞生物基因组测序工作就是针对人类牙菌斑(human tooth plaque)上的细菌开展的。最近几年已经发表了十几篇有关不能人工培养的单细胞生物基因组方面的论文,随着单细胞研究技术与测序技术的进一步发展,我们相信这方面的工作会以指数扩增形式迅速发展起来。随着这些数据的不断积累,我们也会陆续发现更多新的、以前未知的微生物功能和微生物代谢产物(metabolites),发现更多与人类身体健康相关的新物种,甚至有可能彻底改变生命之树的结构,颠覆真核生物、细菌和古细菌之间传统的进化学关系。
微生物在形态学(morphology)、生理学(physiology)和基因型(genotype)方面的多样性也给单细胞分析技术带来了不小的挑战。在我们选择单细胞分析技术、试验反应和化学试剂时,需要考虑每一种样品的特殊性。比如,微生物试验经常需要非常严格的裂解条件,而且不同的微生物往往需要不同的试验条件,这就会增加试验操作的复杂程度。由于在核酸扩增之前并不一定需要进行 DNA纯化操作,所以扩增试剂就需要能够与细胞裂解试剂兼容。复杂的裂解及扩增操作流程比较适合微孔板试验和需要用到整合技术的微流体设备的试验操作,因为这些操作都可以实现自动化。有意思的是,当反应体系缩小到纳升(nanoliters)时,生化扩增仪的表现反而会更好。相对简便的操作规程比较适合反相乳液液滴系统(reverse emulsion liquid-droplet systems)试验,使用这种系统可以快速地进行数万个独立的微反应。到目前为止,几乎所有的单细胞微生物测序结果全都使用了同一个全基因组扩增反应,即多重置换扩增技术(multiple displacement amplification, MDA)。该技术是一种等温的扩增技术(isothermal amplification),使用随机引物,主要依赖的是ɸ29 DNA聚合酶的链置换功能。
3.2 人类单体型(human haplotypes)研究
人类基因组分析工作已经从确定所有人的 “平均”参考序列(reference sequence)快速进入个体基因组测序时代,看起来单细胞技术似乎也帮不上太多的忙。但是我们人类基因组中有一些部分使用传统技术进行分析还是有比较大的难度的。比如人体内的每一个细胞里都含有两套基因组,其中一套来自父亲,另外一套来自母亲,这就叫做单体型现象,而每一个单倍体基因组(haploid genome)中的变异都会对基因的表达、蛋白质的功能,以及疾病造成非常大的影响。
人白细胞抗原( human leukocyte antigen, HLA)编码基因变异就是非常典型的例子, HLA基因单体型信息是骨髓移植工作中非常重要的一项信息,不过这只适用于非常复杂的杂合突变(heterozygous mutation)——在一个基因位点上发生了两个突变。如果这两个突变都位于同一个单体(一条染色体),那么可能是无害的,但是如果分别位于两个不同的单体,那么就极有可能是有害的。现有的技术还无法在基因组的层面上进行这种单体区分(haplotype determination)。传统的、进行单体区分最精确的方法需要对一个家系( family pedigree)进行测序,主要是对父母进行测序。很明显,在临床上大规模开展这种工作是不现实的。
不过单细胞染色体分离技术(Single-cell chromosome isolation)帮了我们的大忙,这是第一种全基因组单体型测量技术(genome-wide haplotype measurement),能够对完整的染色体进行单体鉴定。该技术出现之后很快就与其它技术搭配起来,比如只需要用到少量细胞(不过对于男性精子细胞来说可能需要的细胞数量会多一些)的单细胞测序技术(single-cell sequencing approach)等。我们希望这些技术,以及确定基因组片段单体型的长读长测序技术(long-read sequencing technologies)能够得到更进一步的应用,以促进我们对人类基因组的认识和了解。HLA编码区是我们人类基因组中多态性最明显的一个区域,该区域与人类免疫系统关系密切,也与多种人类疾病有非常直接的联系,所以一直都是研究的热点。不过由于HLA的单体型太过复杂,所以迄今为止也只对少数几个人的HLA区域进行过单体型测序。
单细胞基因组研究工作涉及的另外一个领域就是对各种人的重组方式(recombination pattern)的研究。所谓重组指的是精子细胞和卵子细胞内分别遗传自父系和母系的两条染色体大片段各自断裂,然后相互再连接,形成一个全新的基因组的过程,这也是造成人类遗传多样性的最主要原因。我们知道,整个基因组内的重组几率并不是完全一样的,即存在所谓的“重组热点”,这些位置发生重组的几率要比基因组内其它区域更高。单细胞基因组分析工作的最早成果之一就是发现在不同的个体之间,这些重组热点也会有所差异,这些热点对于某些人而言的确是热点,但是对于另外一部分人来说其实也不是那么热。最近,单细胞研究技术已经被用于分析全基因组重组模式
(genome-wide recombination pattern),以及单个精子细胞的突变率等,世界上也有了第一个针对不同个体的全基因组热点行为研究(genome-wide hot-spot behavior)。我们希望未来的单个精子细胞基因组研究也能够涉及重组突变(recombination mutant),比如对携带罕见PRDM9等位基因的个体开展研究;以及针对与不孕不育疾病(sterility and infertility)相关的、可用于临床诊断的减数分裂功能紊乱(meiotic dysfunction)的研究。
3.3 体细胞突变研究
越来越的人开始慢慢认识到个体基因组测序的意义和价值,不过目前的个体基因组序列指的还是人体内所有细胞基因组的“平均”序列。科学家们在几十年之前就已经发现,人体某些(种)细胞之间是存在基因组差异的,比如属于我们人体免疫系统的B淋巴细胞就是一个很好的例子。每一种B细胞都会严格表达一种特定的抗体,这些B细胞基因组里的基因是绝对不会被重编程(reprogram)的。正如前面已经介绍过的,生殖细胞在减数分裂和遗传重组的过程中也会出现分化和差异。在细胞不断的分裂过程中,以及在可移动的遗传元件(mobile genetic elements)的转移过程中也会慢慢积累各种突变,这些突变都具有非常重要的意义,不过我们目前对此了解得还不是特别清楚。
这些不断积累的突变与衰老,尤其是与肿瘤有非常密切的关系,所以衰老和肿瘤这两个研究领域一定会是单细胞基因组分析技术大显身手的舞台。到目前为止,已经有科研人员利用单细胞研究技术对人体精子细胞和永生化细胞系细胞进行过研究,他们直接检测了这些细胞的自发突变速率(de novo mutation rate)。还有人用这些技术对造血干细胞进行检测,以确定这些造血干细胞的突变程度,判断正常的造血干细胞转化成急性髓性白血病(acute myelogenous leukemia)肿瘤细胞之后的突变程度是不是有了一个大幅度的提升,并借此了解这些白血病肿瘤细胞的演变规律,判断乳腺癌细胞的克隆结构(clonal structure)等。
在成体神经组织里也存在嵌合型突变(Mosaic variation),这些突变与阿尔茨海默病(Alzheimer's disease)等神经退行性病变有关。最近,有科研人员利用单细胞MDA等基因组分析技术在诱导性多潜能干细胞(induced pluripotent stem cell)分化的神经细胞和尸检获得的脑细胞(postmortem brain cell)中发现了大段的(达到 MB级别的)基因拷贝变异(copy number variation)。也有人利用单细胞MDA技术和以PCR为基础的全基因组扩增技术发现了 L1逆转座子(retrotransposition)是促使大脑细胞内出现体细胞嵌合突变的潜在因素,而且还用这种方法发现在不到1/3的脑细胞里存在的突变也同样能够诱发严重的疾病,比如半侧巨脑症(hemimegalencephaly)等。荧光原位杂交技术(fluorescence in situ hybridization)也被用来研究小鼠大脑中部分非整倍体(aneuploid)的神经元细胞与小鼠衰老之间的关系。这是一个让人着迷的研究领域,有各种证据表明嵌合型体细胞突变与机体发育相关,也具有一定的功能,在正常的成熟神经组织里一样能够发现这些突变。这可能就是“正常的”神经表型之间的差异能够导致神经疾病的原因,这些差异也可能与心理疾病相关,而且突变会随着年龄增长越来越多。
3.4 何时需要单细胞测序
什么时候进入单细胞基因组测序项目才合算呢?肿瘤基因组是一种高度异质性的核酸,而且突变的速度非常快,所以对肿瘤组织进行单细胞基因组测序是最合适的。虽然大批量的肿瘤组织测序并没有让科研人员们清楚地认清肿瘤组织的亚克隆组成情况,可是如果再使用单细胞基因组测序技术,我们就可以获得更详细的信息,明确基因组中核酸序列存在高度异质性情况的基因组位点。这种分阶段的技术极大地降低了测序成本,因而增加了对某个肿瘤组织进行测序时可以进行单细胞测序的细胞数目和测序次数。
虽然目前我们还不能确定,对某个肿瘤组织进行多次、大量的单细胞全基因组测序在经济上是否划算,但是对基因组中的重要部分进行分析,或者用测序深度较浅的方法(shallow sequencing)进行低分辨率测序,了解细胞里的基因拷贝数变异情况,也能够得到同样的结果。其实Bridges在80年前开展果蝇基因组研究时就是这么干的。还有一种办法可以代替这种分阶段策略,而且只需一步,那就是对多个单细胞进行全外显子组测序,这样一方面能够了解到肿瘤组织的“总体”外显子组情况,另外也可以发现肿瘤组织内部的亚克隆组成情况,而且成本要比全肿瘤测序(whole-tumor sequencing)经济得多。
3.5 植入前测序
单细胞测序有时是我们发现罕见、或独特细胞的唯一手段。胚胎植入前遗传诊断(Preimplantation genetic diagnosis, PGD)是接受体外受精(in vitro fertilization)等人工辅助生殖技术帮助的夫妻常用的一项技术,在胚胎被植入母体之前,医生们会从体外培养的胚胎中提取一个细胞,对其进行基因组分析。不过对之前开展的临床试验进行荟萃分析(meta analyses)发现,PGD并不是筛查遗传疾病的有效手段,因为在随机对照实验中发现,许多更先进的技术成功率更高,而且生出孩子的几率几乎会高出一倍。应用微阵列比较基因组杂交(array comparative genomic hybridization)等全基因组分析方法可以在胚胎植入前以更高的分辨率对胚胎的基因组进行检测。我们希望这些更高分辨率的基因组分析技术能够尽快应用到PGD临床实践工作当中,能够对胚胎进行结构异常、甚至是点突变的检测。所得的这些数据就可以帮助临床医生们进行更加精细的判断,以了解哪一些胚胎更加健康,可以生下一个健康可爱的宝宝。
3.6 单细胞技术的未来
测序的成本肯定还会不断地降下去。近十年来也诞生了很多生化DNA扩增技术,而且现在又出现了多种单细胞试验手段。然而,目前还没有哪一种核酸扩增技术是绝对的赢家,如果真的出现这样一种技术,那对所有人都会是个意外的大惊喜。但是很难说哪一种核酸扩增技术是最好的技术,因为有很多参数需要考虑。尤其是以下这几点,比如样本类型、反应方式、方便程度(恒温反应还是变温反应,一步法还是多步法)、成本(商品化的还是自制的)、可靠程度(脱靶情况、污染品扩增、扩增时的均一性和误差、扩增技术的覆盖度、错误率,以及嵌合等人工误差)以及最终的得率等。
另外,在比较这些不同的扩增技术时,一定要使用在统计学上相关样品的单细胞样品进行评价,而且一定要避免反应体积、反应方式、裂解条件、污染、样品特异性的差异和细胞间的随机差异所带来的影响。因此,只有针对这些因素做好对照才能找出最好的扩增技术。
另外,还需要开发出自动化的单细胞分离和基因组扩增技术。现有的技术能够处理数百个数量级为单位的细胞,我们可以使用商业化的细胞分选仪完成细胞分选工作,也可以用机械手完成细胞裂解和核酸扩增反应,还可以用微流体设备自动完成上面这一整套操作。自动化和小型化是未来单细胞测序仪的发展方向,这是因为只有分析足够多的样本才能够充分认识样品里的遗传多样性。我们希望芯片技术、微流体技术,以及微型零件加工制造技术(microfabricated approach)都能够有创新性的发展。这样将会极大地提高处理的通量,同时也能够大幅度降低测试成本(降低几个数量级),还可以简化反应步骤,如此便可以在一次实验中对数万个细胞进行单细胞分析。我们相信这只是个时间问题。
单细胞基因组分析技术实际上是多项技术共同发展的结果,而且涉及了生命科学领域里多个基础领域,这将有助于我们解决生命科学领域里的多个重大问题。我们希望随着核酸扩增技术和反应类型的不断发展和多样化,单细胞测序技术的影响力能够进一步扩大,应用到更多的领域,以帮助我们更好地认识和了解整个生命系统。
4. 生物学及医学开始进入单细胞转录组学研究时代
最近的技术进步使得单细胞RNA测序成为了可能。探索性研究已经让我们见识了分化的动态变化过程,细胞对各种刺激做出的反应,以及转录的随机本质。我们正在步入一个单细胞转录组学时代,该研究方向会对生物学和医学产生深刻的影响。
我们现在提到的转录组学(transcriptome)主要源于近二十年来在生物学研究工作中成为主流的群体观测工作(population-level observation)。我们一直习惯于这样一种研究思路,即对整体组织或某个条件下的基因表达倍增情况(明显的或细微的)进行比较,但是细胞之间的实际差异可能会更明显。某些细胞可能会产生非常明显的改变,可是另外一些细胞却“无动于衷”,如此一来,即便那部分发生改变的细胞的变动幅度再大,也会被“沉默的大多数”细胞给掩盖掉和稀释掉。实际上,早在60年前就已经发现,刺激单细胞会得到“全”或“无”这两种截然不同的结果,可如果对一大群细胞进行研究就会得到一个渐进的、可定量式的反应结果。
很明显,对单细胞的基因表达情况进行检测和分析非常有助于我们了解细胞的行为,以及明确都有哪些细胞参与了组织发育、成熟和病变的过程。为了达到这个目的,就需要对单个细胞进行长期的转录组学研究。但是实验技术直到最近才发展到能够对单细胞进行RNA测序的水平,科学家们才能够借助这项技术了解单细胞在基因表达方面有意义的差异。现在也出现了非常详细的实验指南,帮助科研人员构建测序文库,而且FluidigmC1等商业化的单细胞全自动制备系统也极大地降低了广大科研人员涉足这个领域的门槛。单细胞实验操作技术的广泛应用将对我们产生深远的影响,也将帮助我们加深对细胞状态、转录本质以及基因表达调控,乃至对疾病病理进程的认识。
4.1 信噪比问题
单细胞转录组研究工作主要依赖逆转录反应(reversetranscription)。首先,将待研究的RNA逆转录成cDNA,然后再通过PCR反应或体外转录反应(invitrotranscription)进行扩增,最后对扩增产物进行深度测序。不过其中扩增反应非常容易出错,也容易丢失信息。这是由于单个细胞里含有的RNA非常少,所以需要对这些微量的核酸进行大量扩增,以致这个扩增反应产生了大量的偏差。虽然技术噪声会干扰科研人员对低丰度的RNA分子进行高分辨率的测序,但是当前经过改进的实验操作流程已经可以让我们获得足够多的单细胞转录组信息。比如,在单细胞转录组学研究工作中有一个屡次被提及的问题,那就是在未对细胞进行分类的情况下,如何根据细胞的类别或状态对细胞进行准确的、可重复的分类。与细胞类别,或者发育阶段相关的基因表达模式是一个比较可靠的判断依据,远比与细胞周期等动态进程相关的生理变量或者技术噪声值得信赖。另外,有人已经对不同细胞里成百上千个基因的表达差异进行过研究,证实这种单细胞研究技术的确能够发现有意义的信息。最近开展的更深入的研究工作将进一步提高单细胞测序研究的信噪比,因为我们将进一步提高逆转录和PCR反应的效率,也可以采用分子标签(molecular barcoding)策略来控制核酸扩增反应中出现的偏差。
4.2 单细胞转录组学研究工作中存在的挑战
科研人员们基于几种不同的目的开发出了现有的单细胞 RNA测序技术。比如可以对转录子全长序列进行测序,这样就能够了解整个基因和各种转录子亚型(transcript isoform)的序列信息,也有利于我们发现并监测单核苷酸多态性(single-nucleotide polymorphisms)和其它突变的情况。而主要依赖标签,只对转录子5'或3'端进行测序的策略则可以在牺牲全长序列信息的前提下为我们提供与转录子丰度相关的信息,有利于大规模开展分子定量研究。
不过整个单细胞测序界全都在追求同一个目标,那就是用一种经济、高通量的技术对细胞里所有的RNA进行全长序列测序。其中在进行核酸扩增之前如何减少RNA的丢失率,提高RNA逆转录成cDNA的效率是需要重点突破的技术难关,也是有助于提高RNA检测成功率的关键所在。另外一个同样重要的技术就是如何对单细胞进行分离、分类及分选,而且是在不给细胞基因表达情况带来任何扰动的前提下从整块组织中分离得到单个的细胞样品。另外,科研人员们还希望能够在不考虑转录子长度的情况下,同时对poly(A)+ RNA和poly(A)–RNA,以及各种RNA修饰体(比如 m6A)进行检测。
我们现在已经发现,在单细胞测序研究工作中,细胞转录过程有一大特点,会给我们的研究带来非常大的麻烦,那就是我们在对细胞群体的研究工作中发现的细胞基因表达规律在单细胞的水平上其实一点都不可靠,任何随机的扰动都有可能使该基因在某些细胞里不表达,或者表达量很低,但也有可能变得很高。这种多变性可能是因为细胞内的基因表达是一个随机的分子进程,所以在单个细胞内,基因的转录就是一个或全或无的概率性事件。科学家们已经对原核生物和单细胞的真核生物进行过大量的研究,对基因转录的这种随机本质有了非常深刻的认识和了解,现在越来越多的证据表明,哺乳动物细胞内其实也是一样的。因此,我们在开展单细胞转录组学研究工作时也需要注意这一点。比如,标准的基因表达差异试验(differential expression test)可能就不太适于进行单细胞研究,因为在这些被研究的细胞当中,可能有一部分细胞里就没有相应基因的表达。现在已经出现了适于这类研究工作的试验策略,可以将转录子丰度差异与细胞基因表达比例差异结合起来进行观察。
到目前为止,开展单细胞转录组研究时还是需要单细胞悬液(比如组织解离液或者细胞培养悬液)做检测样品,但这种样品不能反映细胞在组织里的空间组织结构信息,除非我们知道这些细胞取自组织的哪一个部位。RNA原位杂交(RNA in situ hybridization)技术可以部分体现这种空间组织结构信息,能够了解组织里某些特定细胞里特定基因的表达情况。不过现在也有人在开发能够同时了解空间结构信息与转录组信息的单细胞研究技术,比如芯片式的多路测序技术(array-based multiplexing strategy)或原位测序技术(in situ sequencing)等。这类技术出现之后将帮助我们了解正在发育中的、成熟之后的、或者病变组织内的单细胞转录组情况,让我们对生命与疾病有更深入的了解,发现转录组与细胞间相互作用、组织极性形成以及局部差异之间的关系。
4.3 单细胞测序技术与生物学的关联
对单个细胞内的基因表达情况进行研究将彻底颠覆我们对基因表达调控的认识和理解,也将解决很多长久以来一直困扰着我们的生物学难题。比如细胞聚集在一起是由细胞种类决定的,还是因为在发育的过程当中,根据细胞的表达谱而决定的。如果是根据细胞基因的表达情况来决定的,那么在对足够多的单细胞进行测序之后我们就可以准确无误地重建出(这也叫反向工程学技术)任何细胞(下图)。如果被研究的细胞数量足够多,而且已经彻底解决了试验误差的问题,那么这种研究就可以发现组织里的所有细胞类型,包括那些尚未被发现的新型细胞。同一个群体的细胞也可以被用来发现特定细胞类型的基因表达谱,此时也一样是以测序结果为依据,也同样在事前不知道哪些组织或细胞里会表达哪种标记基因的前提下。因此,单细胞RNA测序是一种以试验结果为基础的,可以对细胞种类进行定量分析的研究手段。
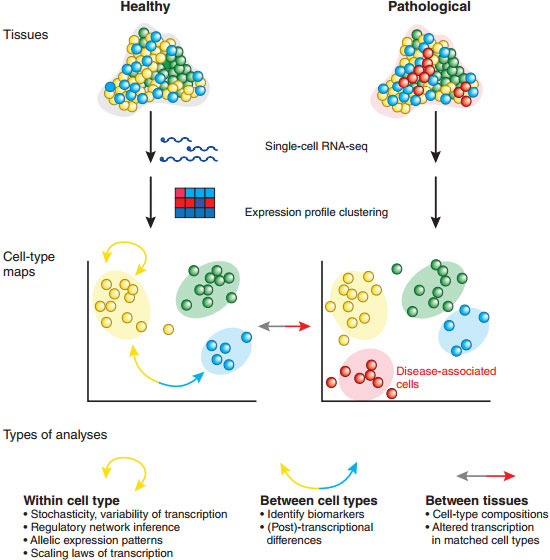
图:对组织和不同的细胞进行单细胞转录组分析。首先将健康组织和病变组织解离,制成单细胞悬液,然后利用单细胞RNA测序技术,以及获得的基因表达谱信息对细胞进行单独的聚类分析,最后可以得到一张细胞聚类分析图。根据此图可以了解组织的细胞构成情况,甚至还可以发现以往不知道的未知细胞。如此丰富的信息还可以用来解决其它生物学问题,比如同种细胞和组织内,或者不同的细胞或组织间的基因表达情况和基因表达调控情况等。
单细胞转录组学研究还可以提供高质量的细胞转录组图谱,这不仅针对稳定的细胞状态,也同时针对复杂多变的细胞状态,比如细胞分化或重编程时的状态。不过要达到这种研究目的,就必须对覆盖整个阶段的、数量足够多的单细胞进行测序研究,这样才能在事后的数据分析工作中重点关注其中的某一个阶段(比如开始出现不同分化方向的那个时刻),得到有价值的研究成果。样品量还反映了我们预计会得到多少细胞种类,或者有多少生物学事件会发生。当然,这也取决于人类基因组当中基因转录的幅度究竟有多大,因为有多个研究发现,人类基因组当中很多基因只发生了很少的转录,平均1万个细胞里只发现了一个转录子拷贝。这种转录子可能是在很少量的细胞里高度表达(比如平均在10万个细胞里有一个细胞内的拷贝数超过了10个),或者是在大量的细胞里都维持非常低的表达水平,即所谓的渗漏表达。对大量的细胞(数千个)进行研究可能会解决这个问题,也有助于我们认识细胞内整体的转录水平和整个细胞的基因表达调控网络。
对人体组织和细胞进行RNA测序分析已经证明,RNA研究手段可以用于各种转录组学及蛋白质组学研究。进行组织比对时发现,大量的差异都是非常细微的,但是发现可变剪接(alternative splicing)情况、多腺苷酸化(polyadenylation)情况和转录起始位点的选择,在单细胞层面上都是一种全(开)或无(关)模式,这也与之前的单细胞研究结果相吻合。针对可变多腺苷酸化调控机制的研究发现,在增殖比较活跃的细胞里,以及体外培养的转化细胞里,转录子3'端非编码区都比较短。单细胞RNA测序技术尤其适用于对体内的肿瘤细胞进行分析,因为针对一堆转化细胞、间质细胞和其它浸润细胞单独提取转录产物进行分析,可以了解各种转录产物的丰度和亚型信息。对离散的肿瘤组织和健康组织进行单细胞转录组分析还可以精确地确定与转化状态相关的、不同的mRNA亚型。
4.4 单细胞测序对于医学的意义
转录组学技术对于医学的意义主要集中在对病变组织和相应健康组织的比较工作上,或者可以对大量的病变组织进行分析,找出其间的差异,即进行亚型鉴定。我们主要是通过细胞组成情况(比如浸润的免疫细胞),以及和转化细胞和周围间质细胞里的基因表达情况来确定肿瘤组织的。因此,在组织层面进行观察时需要同时对几种不同的基因表达谱进行研究。高通量的病变组织单细胞分析能够同时检测细胞的组成变化(通过细胞聚类分析手段)和相应的基因表达变化。我们可以对健康组织和病变组织里特定的细胞进行比较,发现与疾病相关的特异性基因表达改变情况。不过要了解细胞组成成分方面的局部变动信息就一定得在同一块肿瘤组织的不同部位进行多点取样研究。
无法用传统技术开展研究的、比较难获得的、珍贵的临床细胞样品也是能够从单细胞转录组学研究技术中获得好处的一个研究方向。比如数量非常少的循环肿瘤细胞(circulating tumor cell, CTC)就是一个非常好的单细胞研究方向,在一毫升血液中往往也就只有几个 CTC细胞,所以用常规的方法对这些细胞进行全基因组研究几乎是不可能的。两项开创性的研究工作已经证实,可以用单细胞RNA测序技术来判断CTC细胞是黑色素瘤细胞还是前列腺癌细胞。转录谱也已经证实细胞分离步骤没有问题,而且也发现了特异性的基因表达谱变异情况。对CTC细胞的全长转录产物进行单细胞RNA测序在成功检测基因表达情况的同时,可以检测突变情况。对单个CTC细胞进行转录组学分析还是一种无创检测手段,可以帮助临床医生们选择合适的抗癌药物和治疗方案,还可以随时监测病情的进展情况和疗效。现在是时候判断CTC转录组学研究手段对于癌症诊疗工作的指导意义了,也可以根据 CTC细胞上的分子标志物确定将来的靶向治疗方案。
4.5 单细胞测序技术未来的发展前景
由于我们刚刚进入单细胞转录组学研究时代,所以在不久的将来一定会有很多新的发现。RNA丰度与细胞表型(比如细胞大小、核大小等)之间是否存在某种对应关系,这也是一个很有意思的研究课题。比如为了维持不同大小细胞胞膜或者细胞器质膜上的蛋白质浓度,是否需要不同丰度的RNA?基因表达的种类可能与胞膜或核膜的特定区域相关,也可能与胞质体积的大小或核大小相关。只有了解了这些信息之后,我们才可以开始研究细胞的异质性问题,以及组织层面的细胞组成情况等。比如,对由大小不同的细胞组成的两个组织进行比较可能就会发现与细胞大小相关的基因表达特征。对单细胞表达谱进行更深入的研究还可能会为将来的试验设计打下更科学的基础,比如是应该从组织层面、同种细胞,还是单细胞层面,或者综合这三个层面来开展下一步研究。
随着单细胞转录组学研究技术的不断成熟,估计在未来几年内单细胞基因表达及调控研究一定会成为一个新的大热门,科研界很快也会获得足够多(成百上千、甚至是数百万个细胞)的转录子定量研究数据。这些信息可以帮助我们回答很多重要的科学问题,也可以为将来定量研究细胞种类和异质性问题打下基础。根据这些信息,几乎还可以确定复杂的多细胞器官内所有种类细胞的转录组情况。而且单细胞转录组学信息还可以帮助我们提高对基因表达调控网络的人工操纵能力,因为大量的单细胞数据都真实地反映了细胞面临的生物扰动情况,这些信息都能够帮助我们加深对基因表达调控网络的了解。
5. 单细胞测序技术的前景
通常来说,我们会将具有同一表型的细胞看作是一个具有特定功能的整体,并将其称作组织或者器官。不过对单个细胞进行深度DNA和RNA测序会发现,各种各样的细胞状态构成了一个复杂的生态系统,这样一个复杂的系统才形成了组织和器官的整体功能。继续发展高信息度、实时的、多模单细胞检测技术将帮助我们真正认识处于微环境系统下单个细胞的功能。
自从Robert Hooke在1665年时第一次使用“细胞(cell)”这个词来描述他用他自己发明的显微镜观察软木塞时看到的镜下结构之后,细胞就一直是科学家们关注的重点对象。虽然早期的形态学研究(morphological study)已经清楚地确定了各种各样的细胞形态,但是最近的研究还是出乎意料地发现了很多新的、不同的细胞状态(cellstate)。一个标准的人体细胞大约含有60亿个DNA碱基对,以及6亿个碱基的mRNA(这个规模的mRNA已经足以提供超大的编码能力)。对单细胞的DNA和RNA进行深度测序就能够以前所未有的更高的分辨率,更全面地掌握细胞的功能。科学家们对细胞状态的这种特异性识别能力有助于我们更好地了解细胞的正常功能和异常情况。
单细胞测序能够以更高的分辨率发现细胞之间的差异,这也引出了一系列的新问题。其中最根本的问题可能就是发现并衡量出这种细胞间的差异并不一定有意义,也就是说,我们并不知道哪种细胞状态才是真正有功能的细胞状态。由于在一个典型的人体细胞里,每一种mRNA平均大约只存在几十个拷贝,这么少的mRNA分子能够像我们在发育初期看到的那样,对细胞进行精细的调控吗?单个细胞彼此之间又是通过何种相互作用,实现组织层面的功能,这种对细胞生态学(cellularecology)的本质研究是一个非常值得深入挖掘的崭新领域。另外,如果我们认定细胞的表型就是多个细胞所形成的一个局部生态系统的功能,那么,在一个多细胞组成的组织里,那么多的局部生态系统是如何共存在一起的,它们彼此之间有相互交换作用吗(图1)?
虽然单细胞测序技术(single-cell sequencing)给我们带来了很多惊喜,我们对该技术也寄予了厚望,但是该技术目前还不是实验室里一项常规的检测技术。因为基础技术以及数据分析和解读技术的不断进步是提高单细胞测序技术精确度的关键,而要在系统层面了解单细胞的作用,则必须要对大批量的细胞进行单细胞测序研究。我们接下来会对这些问题进行评述,同时也会重点介绍单细胞测序技术未来的发展方向,以及新近出现的单细胞测序补充技术,还将介绍单细胞在整个生态大环境下的具体功能。
5.1 关于单细胞研究的几个重要问题
有多个重要的问题会影响单细胞测序研究所获得的数据质量。其中尤其需要注意的、不可避免的问题就是,转录组(transcriptome)会根据各种刺激做出改变,而且这种改变在单细胞层面上表现得更加突出。考虑到这一点,我们应该慎重对待单细胞转录组数据,(至少在一定程度上)将其看作是干扰试验(perturbationexperiment)的结果,除非能够开发出破坏性较低的RNA分离技术。
5.1.1 细胞分离问题
单细胞分离技术几乎算是最需要开发,也最需要建立一套标准化体系的技术。使用膜片钳(patch pipette)或纳米管(nanotube)获取单个细胞的胞质内容物是目前分离细胞RNA的常规方法,但是这种操作容易遗漏细胞器成分。使用微流体设备(microfluidic device)可以分离得到一个个单独反应室里的细胞,但是需要将细胞与其它底物分离开,而这些底物有可能会干扰细胞的转录状态。细胞在解离、分类富集的过程中,细胞的转录状态是否发生改变,就是要特别注意的一个问题。分散培养的细胞非常容易分离,但是用这种细胞做实验需要非常好的试验设计,以免因为缺乏微环境的影响而造成实验结果解读问题。最理想的情况是在组织、或者天然的微环境状态下,对单细胞进行内容物分离操作。只有这样,进行单细胞mRNA检测才能够反映出细胞在整体条件下最真实的状态,也只有这样,才能尽可能地减少人为操作给细胞带来的影响。
5.1.2 核酸扩增问题
在缺乏成熟的、强大的单分子测序技术的情况下,开展单细胞研究最大的问题就是底物(核酸)的扩增问题,因为扩增失误往往会导致最终的测序结果发生偏差,让我们无法得到目标核酸的序列。进行DNA测序时这个问题显得尤为突出,因为只有一个DNA分子可供测序。DNA测序的最大问题就是测序的覆盖度(coverage)问题。以PCR技术为基础的扩增技术能够获得很高的覆盖度,但是会带来扩增不均一(uneven)和错误扩增的问题。如果要进行错误修正(error correction),并发现单碱基突变(single nucleotide variant),这又需要额外的统计学方法。对于单细胞测序而言,错误修正更加困难,因为缺乏好的对照,而且我们根本不知道单个细胞之间究竟会有多少个变异。
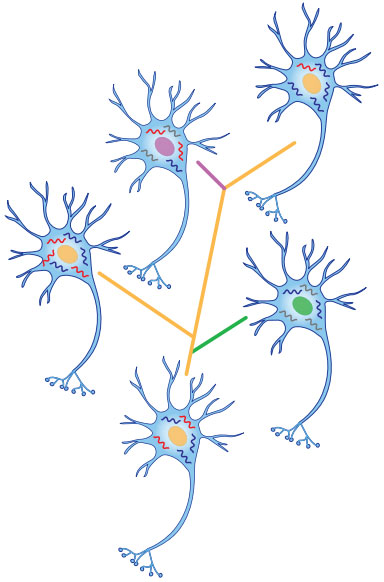
图:经过单细胞测序发现的多种不同的单细胞状态。图中“核”表示DNA,“折线”表示RNA,每一种不同的颜色代表这些核酸拥有不同的序列。从图中可以看出,这些看起来一样,或者彼此相近的细胞其实在核酸序列水平上是不一样的。
对于RNA分子而言,最大的问题是如何在扩增的过程中保证这些分子之间最初的(丰度)比例关系。RNA扩增的第一步就是利用逆转录酶(reverse transcriptase, RT)获得互补DNA(complementary DNA, cDNA)。 在单细胞转录组测序工作中,这是最关键的一个步骤,RT反应的效率直接决定了细胞里最终有多少RNA能够被测序。RT酶最初来自感染了小核糖核酸病毒(picornavirus)的哺乳动物细胞,这种酶的效率非常高,细胞里哪怕只有一个病毒RNA拷贝,也能够合成出全长病毒核酸。虽然这种RT酶在体外实验中没有表现出太强的持续合成能力(合成出全长产物的几率还不到10%),但是经过优化之后,其持续合成能力也能够达到90%。突变之后的RT能够合成出更长的cDNA产物,在RNA浓度不佳时这种突变RT酶更加适用。
单细胞PCR技术(single-cell PCR)能够让这些源自RNA的cDNA分子以指数形式扩增。虽然在很多研究中都会使用PCR技术来构建测序文库,但是我们也应该清楚,PCR针对某些特定序列(比如高GC含量或茎环结构等)的低反应效率也会呈指数形式扩增。所以大部分科研人员都会尽量减少PCR的反应循环数,就是为了减少这方面造成的误差。不过由于这种扩增误差主要源自特定的序列,而且基因的表达水平本来就千变万化,所以我们很难估计误差究竟有多大。虽然某些序列的转录效率也不是太高,而且会生成较短的扩增产物或者缺失某些序列,但是以cDNA体外转录技术(in vitro transcription of cDNA into amplified RNA, aRNA)为基础的线性扩增技术(Linear amplification)还是能够在一定程度上解决这种因为扩增而带来的误差问题。如果我们的研究目的只是对RNA进行定量,而不是研究间接突变体,那么生成较短的RNA转录产物问题还不算太大。对经过系列稀释的对照转录RNA进行测序,同时对测序结果进行泊松分布(Poisson distribution)分析,结果证明,这种aRNA扩增方法的分辨率能够达到对2至4个分子进行定量分析,不过试验结果也会受扩增和回收效率的影响。
有一种解决这种扩增偏差的策略是在 cDNA第一链合成时掺入特定的分子信标(sequence tag)。由于我们有大量的分子标签可供选择,所以源自每一个RNA分子的每一个cDNA分子都可以带上独特的标签。在 PCR扩增时,其偏差不会影响这些标签分子(除非标签分子失落),所以就不会造成扩增偏差问题,标签分子的数量就可以准确地反映出细胞里原始RNA分子的数量。不过这种标记技术还非常复杂,目前仍在优化当中。
5.1.3 动态范围和细胞数问题
目前估计,在一个典型的哺乳动物细胞内,大约有5000至15000个不同的基因在转录和表达。如果我们认为每一个基因的情况都是不同的,那么要确定转录组的协方差(covariance),理想状态应该是比自由度(degrees of freedom)多检测10至30倍。如果这些基因之间的变化情况是非线性的,而且更加复杂,那么检测的次数还应该更多。目前没人知道单细胞转录组的自由度究竟有多大,但是至少会有数千个,这说明至少需要对数万个细胞进行测序。现在已经有这样规模的研究工作正在进行当中,不过只针对少数几个特定的靶分子,而且测序的覆盖度也很低。因此,在研究单细胞转录组时,如果要获得足够的转录组覆盖度,需要对多少个细胞进行测序研究,这也是一个非常重要的课题。
多个研究认为,细胞表达量最高的基因平均大约有3000至5000个转录产物。但是通过查阅文献,以及我们实验室自己的经验,我们发现在细胞内,大约90%的转录组产物都不到50个分子。这就产生了一个问题,这么低的表达量能够决定细胞的表型和功能吗?我们都知道,很多基因都存在“开”和“关”这两种状态,而且这些基因的开关状态在一群细胞里是不一样的,另外还有很多表达水平很低的基因,在组织学研究工作中是根本发现不了的。在这些一个细胞里转录子含量还不到50个分子的基因的补体中有很多非常重要的因子,比如转录因子和信号转导分子等。所以我们不能忽视敏感性(sensitivity)问题,而且充分覆盖每一个转录组的动态范围(dynamic range)与对足够数量的细胞进行测序具有同等重要的意义。
5.2 空间问题
荧光原位杂交技术(fluorescence in situ hybridization, FISH)也是研究细胞内RNA分子的一项技术。目前FISH技术通常都会使用多种短片段荧光标记探针,这些小分子探针能够自如地进入组织和细胞内部,与目标RNA片段结合。由于FISH技术在敏感性方面有非常大的改进,所以很难像芯片那样进行选择性杂交,我们也不知道在细胞交联之后,有多少 RNA可用于杂交试验。更重要的是,不能同时对这些发射广谱各异的、数量有限的荧光分子进行“转录组式”的检测(即同时对各种 RNA分子进行检测)。据报道,现在可以同时对细胞内大约30种不同的mRNA(荧光探针)分子进行检测,这和以往的FISH技术相比已经是相当大的进步了,不过这还不够。
多个研究小组都在开发原位测序技术(in situ sequencing),以及组合式标记技术,但是即便细胞内所有的RNA分子在空间上都是等距的,我们现有的显微分辨率(一个标准的 20 × 20微米的哺乳动物细胞组织切片在250纳米分辨率的光学显微镜下)最多也只能分辨大约13000个色点(像素,每一个像素代表一个RNA),而一个细胞内至少有10万至30万个mRNA分子。不过这种对细胞内RNA分子空间分布情况的研究也有助于我们认识细胞的功能和表型。
5.3 单细胞蛋白质组学研究
科学家们往往借助对转录组的研究来了解细胞内的蛋白质组学情况。目前也不是十分清楚细胞内mRNA与蛋白质丰度之间的关系,因此急需一种能够直接评价转录组与功能蛋白质组之间关系的技术。蛋白质复杂的化学特性让我们很难像对RNA进行定量研究那样对蛋白质进行精确的定量研究,不过随着质谱技术(mass spectrometry)的灵敏度变得越来越高,蛋白质挥发技术(volatize)越来越成熟,我们也看到了进行单细胞蛋白质研究的希望。另外,由于高亲和力抗体、纳米体(nanobody)、抗体单链可变区片段(single-chain variable fragment)等抗体衍生物,以及配体(aptamer)的不断发展,这些高亲和力技术也能够给我们提供灵敏性更高的技术手段,让单细胞蛋白质组学研究早日变成现实。
除了测序之外,我们还需要进行其它方面的单细胞研究,比如单细胞 DNA结构研究和单细胞表观基因组学(epigenome)研究等。染色体构象( Chromosomal conformation)、 DNA甲基化( DNA methylation)、染色质结构打开以及小分子代谢组学(small-molecule metabolome)等技术也都在朝着单细胞层面迈进。不论是何种细胞,对组织里的活细胞进行实时的、多变量、多维度的检测才是我们最需要的理想检测手段,因为只有这样才能获得最真实的、最系统的细胞状态和数据。对于RNA分子而言,这可能就意味着活细胞单个转录子分子的检测。这种检测不仅能够发现都有哪些分子参与每一个生物学进程,而且也能帮助我们对每一个生物学进程有更深入的了解和认识。
除了检测和分析之外,我们还需要在单细胞水平进行一些干扰实验,以便对细胞的功能有一个动态的了解。比如使用 RNA分子来调节细胞的功能,甚至还能够起到治疗的效果。用定量稀释的 RNA转染细胞是第一个被报道的转录组诱导的表型重构技术(transcriptome induced phenotype remodeling, TIPeR)。全转录组,或者部分 RNA分子被转染进细胞之后,能够让细胞的表型朝着既定的目标发生变化。TIPeR技术的目标就是利用细胞的“RNA记忆”来实现特定的细胞功能,这是一种能够调节细胞功能和表型的功能基因组学技术。转录组分析和定量调控技术让我们能够操纵细胞的表型和功能,这不论是对于基础科学研究还是临床治疗都有非常重要的意义。
5.4 单细胞生物学研究的前景
在单细胞层面,所有的疾病在病理学上都是不一样的。单细胞研究能够帮助我们更好地认识为什么有些细胞生病了,而另外一些细胞却还是正常的;也能够告诉我们为什么有些细胞对药物的敏感性非常高,可是另外一些细胞对药物却“无动于衷”。科学家们已经发现了很多受疾病影响最明显的细胞或组织特点,以及与疾病发病或严重程度有关的细胞或组织特点。找出这些与疾病相关的特异性分子状态有助于我们发现,并很好地利用药物作用靶点,但是能否发现这些靶点却取决于我们能否很好地认识“生病的”细胞。
比如,我们都知道多巴胺能的神经元细胞(dopaminergic neuron)在患者患上帕金森氏病之后会逐渐失去合成并分泌多巴胺(dopamine)的能力,而且这些细胞最终都会随着病情的进展逐渐死亡。在这些神经元细胞上发现的每一个受体、离子通道蛋白或者转运体蛋白都可以是药物作用的靶点,可以延缓病情的进展或者改善患者的病情。现在用来治疗帕金森氏病的药物主要针对这些神经元细胞上的四种蛋白,它们分别是多巴胺转运体蛋白(Dopa transporter)、毒蕈碱性受体M1(muscarinic receptor M1)、单胺氧化酶(monoamine oxidase, MAO)以及腺苷A2A受体(adenosine A2A receptor)。之前的组织学研究已经发现了药物作用靶点,可是其中有很多靶点都不在目标细胞上。单细胞研究特有的敏感性和特异性告诉我们,在多种细胞上至少存在300至400种不同的药物作用基因。如果帕金森氏病患者也是如此,那么在漫长的疾病进展期内,我们至少可以选择30至40种药物作用靶点进行有针对性的治疗。
除了转化研究作用之外,单细胞研究还能够从根本上改变我们对多细胞组织(器官)工作方式的看法,让我们提出很多新的科学问题。比如在人体数千亿个细胞当中,究竟有多少种不同的细胞?体细胞DNA变异对于细胞的鉴定和细胞的多样性究竟意味着什么?如果体细胞突变非常常见,那么它们是随机发生的,还是属于基因组有计划变异的一部分?细胞的表型是由其自身基因组决定的,还是周围环境动态影响的结果?换句话来说就是,DNA是执行程序的执行者还是只是一个信息的载体而已?
微生物组测序数据不断表明,单细胞微生物就是一个多细胞宿主的组成部分。另外一方面,对一个多细胞组织里的单个细胞进行DNA和RNA测序研究也发现,这些细胞具有极大的异质性。这说明多细胞生物里的细胞并不像每一个生物体内的组织那样没有明显的差异,这些组织的功能是由这些细胞组成的生态系统决定的,而这些细胞彼此之间的相互作用决定了整个组织的表型,这种情况与微生物组非常类似。如果这是所有生物的共同准则,那么确定单细胞的多样性,以及细胞之间的生态系统将是我们认识每一个生物的必然途径。
原文检索:
Kelly Rae Chi. Singled out for sequencing. Nature Methods, 30 December 2013; doi:10.1038/nmeth.2768
Tal Nawy. Single-cell sequencing. Nature Methods, 30 December 2013; doi:10.1038/nmeth.2771
Paul C Blainey & Stephen R Quake. Dissecting genomic diversity, one cell at a time. Nature Methods, 30 December 2013; doi:10.1038/nmeth.2783
Rickard Sandberg. Entering the era of single-cell transcriptomics in biology and medicine. Nature Methods, 30 December 2013; doi:10.1038/nmeth.2764
James Eberwine, Jai-Yoon Sul, Tamas Bartfai & Junhyong Kim. The promise of single-cell sequencing. Nature Methods, 30 December 2013; doi:10.1038/nmeth.2769
原文来自:http://www.bio360.net/news/show/9603.html
1F
恩~
2F
真的好长 :razz: