

批次效应(batch effect)简单说来就是因为实验做了几个批次导致的实验误差,比如芯片数据,每次都要用机器读取,那么光照时间和强度每次都可能不一样, 极有可能出现批次效应。
再比如,实验的三个重复,时间有间隔,也会有批次效应,做过实验的人都知道,Western blot的三个重复那个大bar会戳死人的。
假如解决了这个批次问题,不仅可以让实验更可靠,更厉害的是,我们可以做多个芯片的联合分析了。
我们一般使用sva的中combat包来校正批次效应,有一篇文章比较了6种方法,最后说caombat最好。
下面是举例子: 安装必要的R包并加载,comat就在sva包中。
BiocInstaller::biocLite("sva")
BiocInstaller::biocLite("bladderbatch")
library(sva)
library(bladderbatch)把数据准备好,这是包里内置的数据集
data(bladderdata) #bladder 的属性是EsetExpressionSet,所以可以用pData和exprs方法 pheno <- pData(bladderEset) # 注释信息 edata <- exprs(bladderEset) # 表达矩阵
再做一个组,用于批次效应中排除项。
pheno$hasCancer <- as.numeric(pheno$cancer == "Cancer")
参看一下pheno里面有54行,4列构成,里面记录了批次信息 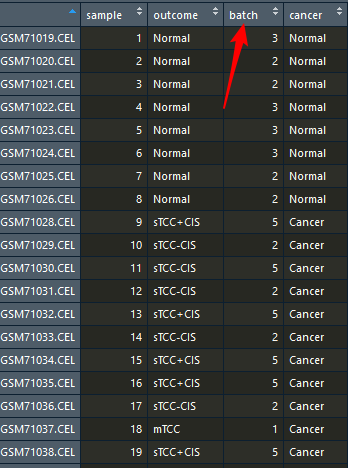
有没有批次效应,眼睛可以看得出来。 使用Hierarchical clustering的方法去看一下聚类的情况
dist_mat <- dist(t(edata)) clustering <- hclust(dist_mat, method = "complete") plot(clustering, labels = pheno$batch) plot(clustering, labels = pheno$cancer)
确实,因为批次的原因,聚类时在正常组中混入了肿瘤,混入的样本批次跟normal中的不一样。 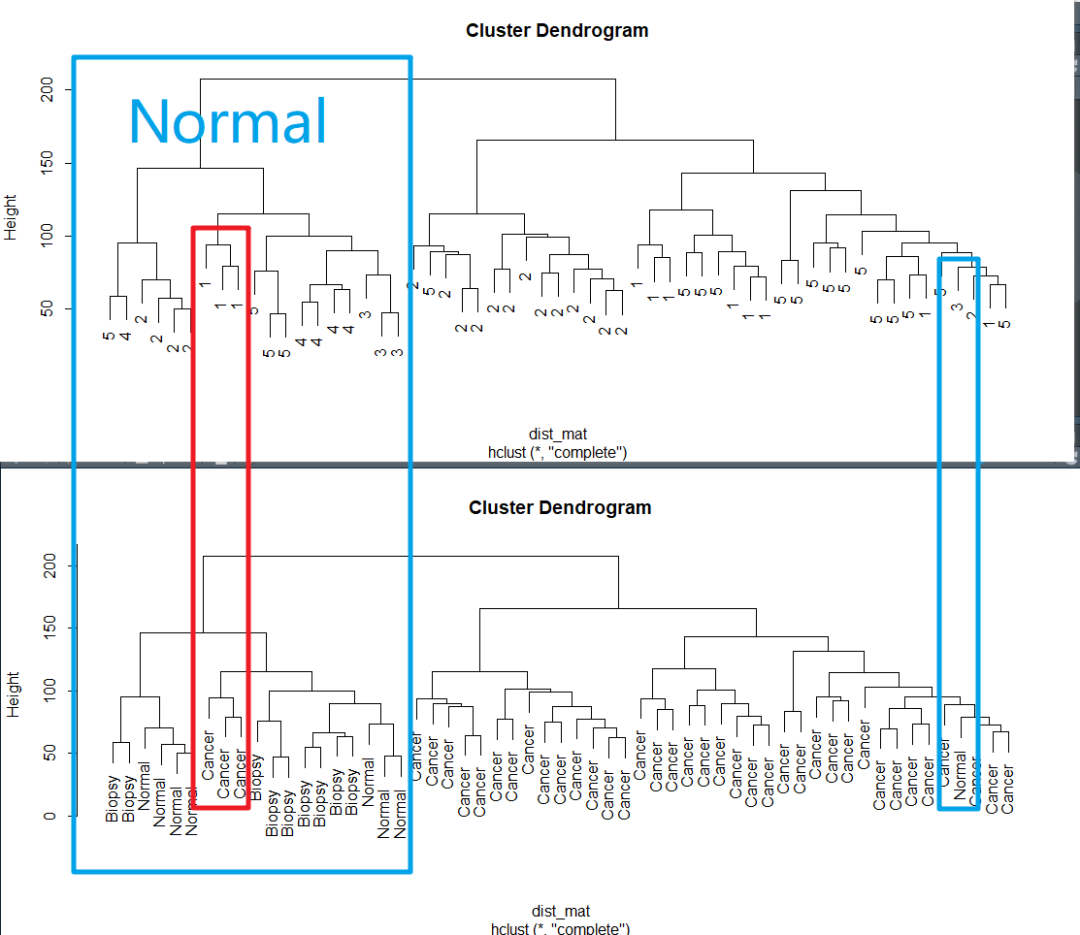
校正批次效应,model可以有也可以没有,如果有,也就是告诉combat,有些分组本来就有差别,不要给我矫枉过正!
model <- model.matrix(~hasCancer, data = pheno) combat_edata <- ComBat(dat = edata, batch = pheno$batch, mod = model)
这时候我们再来画个图看看
dist_mat_combat <- dist(t(combat_edata)) clustering_combat <- hclust(dist_mat_combat, method = "complete") plot(clustering_combat, labels = pheno$batch)
是这个样子的,聚类正常了。 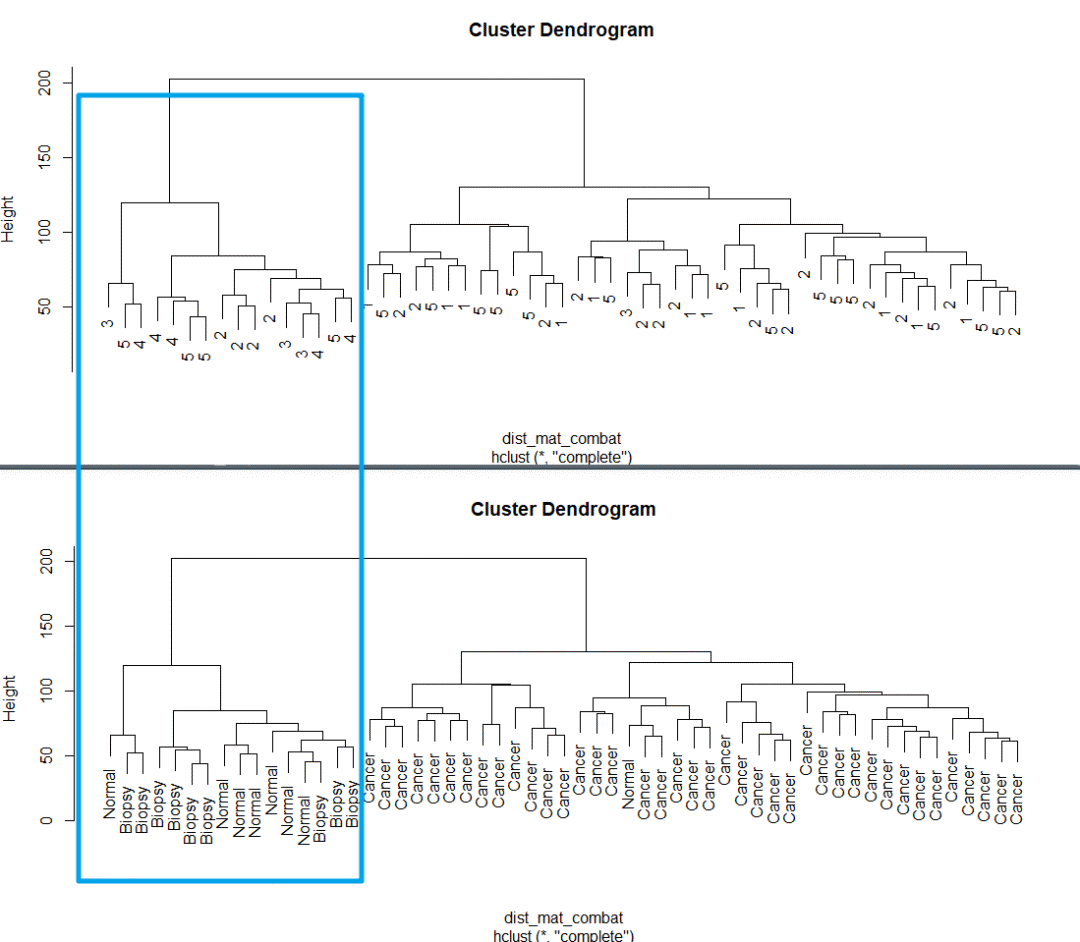
这时候就可以做下游差异分析了。
1F
“我们一般使用sva的中combat包来校正批次效应,有一篇文章比较了6种方法,最后说caombat最好。”这篇文章具体是哪篇呢?
2F
您好,请问用R语言中combat去除批次效应后数据集中会有负值出现,在后续的分析中我不希望有负值,应该怎么处理呢?可以直接加上数据集中最小的负值吗?