

对于RNA-seq而言,由于 技术误差, 测序深度不同, 基因长度不同,为了能够比较不同的样本,比较不同的基因的表达量,以及使表达水品分布符合统计方法的基本假设,就需要对原始数据进行标准化。
对于一个新兴的领域,通常会有50多种算法,但是最后常用的,其实也就那么几个。在RNA-seq标准化这个领域也是如此,目前用的最多也就是, RPKM/FPKM, TPM,但是注意,有些时候一个方法出现的多,单纯是因为公司没有修改他们的分析流程。
为了方便理解,假设目前你在一次测序中(即剔除批次效应)检测了一个物种的3个样本,A,B,C,这个物种有三个基因G1,G2,G3, 基因长度分别为100, 500, 1000. 通过前期数据预处理,你得到了尚未标准化的表达量矩阵,如下所示。
事先声明,以下数字完全是我瞎编,方便用于后续的计算。
| 基因/样本 | 样本A | 样本B | 样本C |
|---|---|---|---|
| G1(100) | 300 | 400 | 500 |
| G2(500) | 700 | 750 | 800 |
| G3(1000) | 1000 | 1300 | 1800 |
基因表达量矩阵
| 基因/样本 | 样本A | 样本B | 样本C |
|---|---|---|---|
| G1(100) | 300 | 400 | 500 |
| G2(500) | 700 | 750 | 800 |
| G3(1000) | 1000 | 1300 | 1800 |
先说三个简单的策略,也就是最容易想到的方法
- Total Count, TC, 每个基因计数除以总比对数, 即文库大小, 然后乘以不同样本的总比对数的均值
- Upper Quartile, UQ, 和TC方法相似, 即用上四分位数替代总比对数
- Median, Med, 和TC方法相似, 用中位数代替总比对数
上面方法都相似,考虑到我的例子只有三个基因,所以只展示TC方法的结果. 可以发现,原本比其他组观测值的A-G2,目前反而是最高。
| TC处理后 | A | B | C |
|---|---|---|---|
| G1 | 370 | 402.7 | 418.1 |
| G2 | 863.3 | 744.1 | 543.5 |
| G3 | 1233.3 | 1308.8 | 1505.1 |
如果省去TC中的 "乘以不同样本的总比对数的均值" 这一步,那么差不多就是CPM (counts per million)的策略,也就是根据直接根据深度对每个样本单独进行标准化. 在edgeR和limma/voom里面都有出现过。
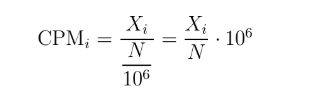
TMM(trimmed mean of M value)方法出现在2010年,比TC、 UQ、Med, CPM方法高级一点,基本假设是绝大数的基因不是差异表达基因.计算方法有点复杂,简单的说就是移除一定百分比的数据后,计算平均值作为缩放因子,对样本进行标准化。这次我们用R/edgeR来算. 和之前不同,A组的G2基因标准化后还是最低,这就是trim所引起。
> library(edgeR)
> expr <- matrix(c(300,400,500,700,750,650,1000,1300,1800),nrow = 3,byrow = TRUE)
> f <- calcNormFactors(expr, method = "TMM")
> mt_norm <- t(t(mt) /f )
> mt_norm
[,1] [,2] [,3]
[1,] 303.0164 402.5300 491.9114
[2,] 707.0382 754.7438 787.0583
[3,] 1010.0545 1308.2226 1770.8811DESeq2/DESeq有自己专门的计算缩放因子(scaling factor)的策略,它的基本假设就是绝大部分的基因表达在处理前后不会有显著性差异,表达量应该相似,据此计算每个基因在所有样本中的几何平均值(geometri mean), 每个样本的各个基因和对应的几何平均数的比值的中位数就是缩放因子(scaling factor). 这里仅仅提到思想,不做计算。
上述方法都是对样本整体进行标准化,标准化的结果只能比较不同样本之间的同一个基因的表达水平。如果要同时比较不同样本不同基因之间的表达量差异,就得考虑到每个基因的转录本长度未必相同,毕竟转录本越长,打算成片段后被观察到的概率会高一点。最长尝试解决这个问题,应该是单端测序时代的RPKM(双端则是FPKM), 全称为Reads Per Kilobase Million, 其中K象征的是转录本长度, M象征的是测序量.

对于A-G1而言,他的表达量就是300*1000 / (2000 x 100) = 1.5, 其中系数10e6在这里不需要使用。你可以认为它是先对文库大小进行标准化,然后再根据基因长度标准化
| RPKM处理后 | A | B | C |
|---|---|---|---|
| G1 | 1.5 | 1.63 | 1.61 |
| G2 | 0.7 | 0.61 | 0.52 |
| G3 | 0.5 | 0.53 | 0.58 |
一波操作下来,感觉数值就有点可比性了。
下面介绍TPM(transcript per million), 计算公式如下,其中X表示比对到基因上的read数,l表示基因的长度。
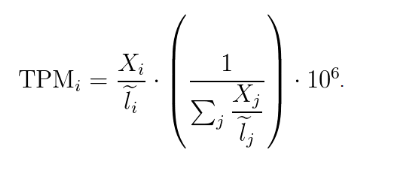
如果手动计算的话,那就是先进行 长度标准化,
| 长度标准化 | A | B | C |
|---|---|---|---|
| G1 | 3 | 4 | 5 |
| G2 | 1.4 | 1.5 | 1.6 |
| G3 | 1 | 1.3 | 1.8 |
然后进行文库标准化,即每一列各个数值除以每一列的和
| 长度标准化 | A | B | C |
|---|---|---|---|
| G1 | 0.56 | 0.59 | 0.60 |
| G2 | 0.26 | 0.22 | 0.19 |
| G3 | 0.18 | 0.19 | 0.21 |
| 列和 | 1 | 1 | 1 |
目前还有一种技术叫做UMI,可以进行转录本的绝对定量,一个转录本有多少特异的UMI,就一定程度上代表了它的表达量。
一些看法
RNA-seq数据标准化其实要分为两种,样本间标准化和样本内标准化。
对于差异表达分析而言,样本内对不同转录本长度进行标准化 毫无必要,是的,真的是没啥意义,粗略比较同一个基因在两个样本间是否有差异,只要处理好测序深度这个问题就行,任何要求用RPKM/FPKM而不是raw count作为差异表达分析的原始数据的分析方法都需要被淘汰掉,而沿用这种分析策略的公司都已经很久没有更新自己流程了。
此外,如果你要比较不同样本内的基因表达情况,那么目前更推荐用TPM。 因为在它的计算方法中,能更有效的标准化不同转录本组成上的差异,而不是简单除以文库大小。
题外话,我最近看到一篇发在Nature上ATAC-seq文章,Method部分提到他用RPKM这个方法对每个bin的read count进行标准化。考虑到每个bin的大小都一样,我觉得这个标准化的方法从定义上更接近CPM。
Correcting for gene length is not necessary when comparing changes in gene expression within the same gene across samples, but it is necessary for correctly ranking gene expression levels within the sample to account for the fact that longer genes accumulate more reads.
对于差异表达分析而言,标准化不但要考虑测序深度的问题,还要考虑到某些表达量超高或者极显著差异表达的基因导致count的分布出现偏倚, 推荐用TMM, DESeq方法进行标准化。
参考文献
- TMM: A scaling normalization method for differential expression analysis of RNA-seq data
- TPM: RNA-Seq gene expression estimation with read mapping uncertainty
- RNA-seq必看综述: A survey of best practices for RNA-seq data analysis
- Estimating number of transcripts from RNA-Seq measurements (and why I believe in paywall)
- What the FPKM? A review of RNA-Seq expression units