

准备工作
参考基因组
测序得到的是几百bp的短read, 相当于把拼图打散了给你。如果没有参考基因组,从头(de novo)组装等于是重走人类基因组计划的老路,也就是打散了拼图,却不告诉你原来是什么样子,那么任务将会及其艰巨。
还好人类基因组已经组装好了,我们只需要把我们测得序列回贴(mapping)回去,毕竟人与人之间的差距只有不到1%差异, 允许mismatch就行。
因此第一步就是要去UCSC(http://genome.ucsc.edu/index.html)下载hg19参考基因组(文献要求)
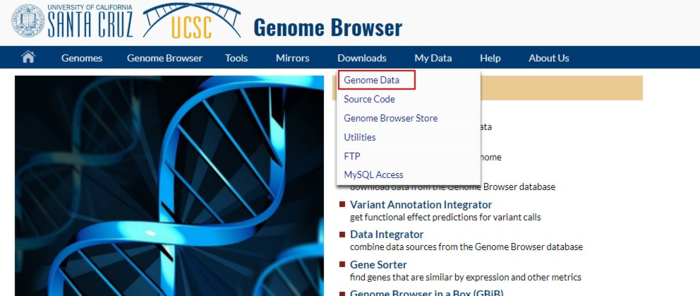
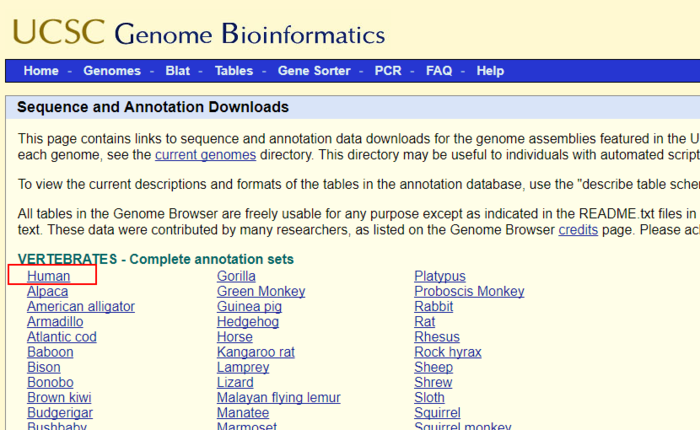
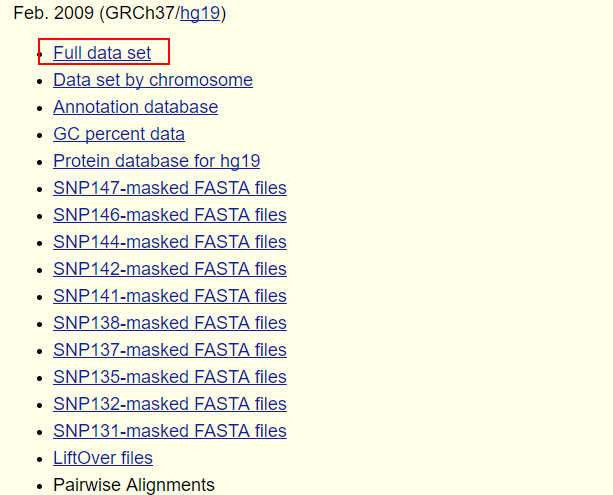
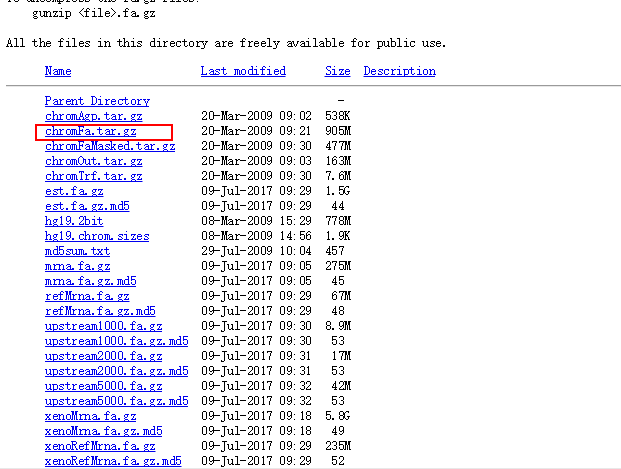
不同文件的所包含的数据在该页面有介绍,其中
chromFa.tar.gz - The assembly sequence in one file per chromosome.Repeats from RepeatMasker and Tandem Repeats Finder (with period of 12 or less) are shown in lower case; non-repeating sequence is shown in upper case.
我将数据存放在Windows的F盘的Data文件夹下,用于后续操作
cd /mnt/f/Data mkdir reference && cd reference mkdir -p genome/hg19 && cd genome/hg19 nohup wget http://hgdownload.soe.ucsc.edu/goldenPath/hg19/bigZips/chromFa.tar.gz & tar -zvf chromFa.tar.gz cat *.fa > hg19.fa rm chr*
这个对新手来说,基因组各种版本需要注意,hg19、GRCH37、 ensembl 75这3种基因组版本应该是大家见得比较多的了,国际通用的人类参考基因组,其实他们储存的是同样的fasta序列,只是分别对应着三种国际生物信息学数据库资源收集存储单位,即NCBI,UCSC及ENSEMBL各自发布的基因组信息而已。有一些参考基因组比较小众,存储的序列也不一样,比如BGI做的炎黄基因组,还有DNA双螺旋结构提出者沃森(Watson)的基因组,还有2016年发表在nature上面的号称最完善的韩国人做的基因组。前期我们先不考虑这些小众基因组,主要就下载hg19和hg38,都是UCSC提供的,虽然hg38相比hg19来说,做了很多改进,优点也不少,但因为目前为止很多注释信息都是针对于hg19的坐标系统来的,我们就都下载了,正好自己探究一下。也顺便下载一个小鼠的最新版参考基因组吧,反正比对也就是睡个觉的功夫,顺便分析一下结果,看看比对率是不是很低。
注释信息
然而参考基因组是一部无字天书,要想解读书中的内容,需要额外的注释信息协助。
因此第二步,就是去gencode数据库(http://www.gencodegenes.org/)下载基因组注释文件。
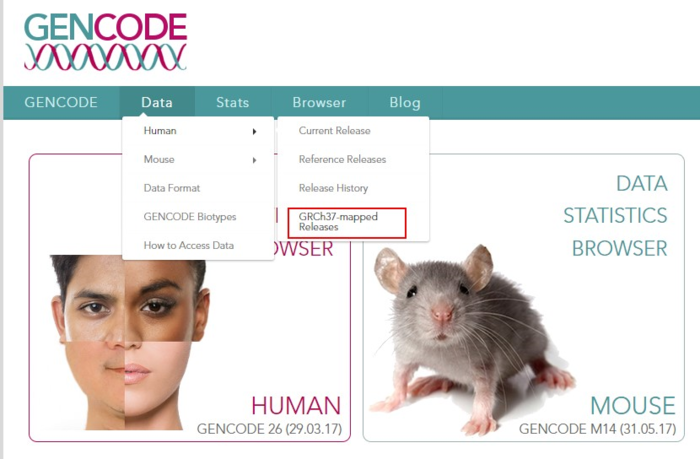

又到了GTF还是GFF3的抉择时刻,简单介绍了一下他们的格式

GTF(General Transfer Format)其实就是GFF2,以Tab分割,分为如下几列
- seqname - name of the chromosome or scaffold; chromosome names can be given with or without the 'chr' prefix. Important note: the seqname must be one used within Ensembl, i.e. a standard chromosome name or an Ensembl identifier such as a scaffold ID, without any additional content such as species or assembly. See the example GFF output below.
- source - name of the program that generated this feature, or the data source (database or project name)
- feature - feature type name, e.g. Gene, Variation, Similarity
- start - Start position of the feature, with sequence numbering starting at 1.
- end - End position of the feature, with sequence numbering starting at 1.
- score - A floating point value.
- strand - defined as + (forward) or - (reverse).
- frame - One of '0', '1' or '2'. '0' indicates that the first base of the feature is the first base of a codon, '1' that the second base is the first base of a codon, and so on..
- attribute - A semicolon-separated list of tag-value pairs, providing additional information about each feature.
而GFF3(General Feature Format)的格式如下
- seqid - name of the chromosome or scaffold; chromosome names can be given with or without the 'chr' prefix. Important note: the seq ID must be one used within Ensembl, i.e. a standard chromosome name or an Ensembl identifier such as a scaffold ID, without any additional content such as species or assembly. See the example GFF output below.
- source - name of the program that generated this feature, or the data source (database or project name)
- type - type of feature. Must be a term or accession from the SOFA sequence ontology
- start - Start position of the feature, with sequence numbering starting at 1.
- end - End position of the feature, with sequence numbering starting at 1.
- score - A floating point value.
- strand - defined as + (forward) or - (reverse).
- phase - One of '0', '1' or '2'. '0' indicates that the first base of the feature is the first base of a codon, '1' that the second base is the first base of a codon, and so on..
- attributes - A semicolon-separated list of tag-value pairs, providing additional information about each feature. Some of these tags are predefined, e.g. ID, Name, Alias, Parent - see the GFF documentation for more details.
看不出来有啥区别,不想纠结就全下载好了。
nohup wget ftp://ftp.sanger.ac.uk/pub/gencode/Gencode_human/release_26/GRCh37_mapping/gencode.v26lift37.annotation.gtf.gz &
nohuop wget ftp://ftp.sanger.ac.uk/pub/gencode/Gencode_human/release_26/GRCh37_mapping/gencode.v26lift37.annotation.gff3.gz &
我们对文字的理解能力远远小于图片,所以下一步需要下载基因组浏览器
IGV, Integrative Genomics Viewer
下载地址为: http://software.broadinstitute.org/software/igv/download
Windows下载如下版本, 会自带一个java运行环境
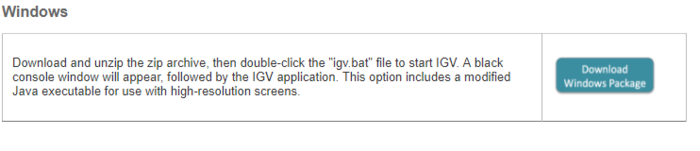
双击igv.bat, 就会出现运行界面。
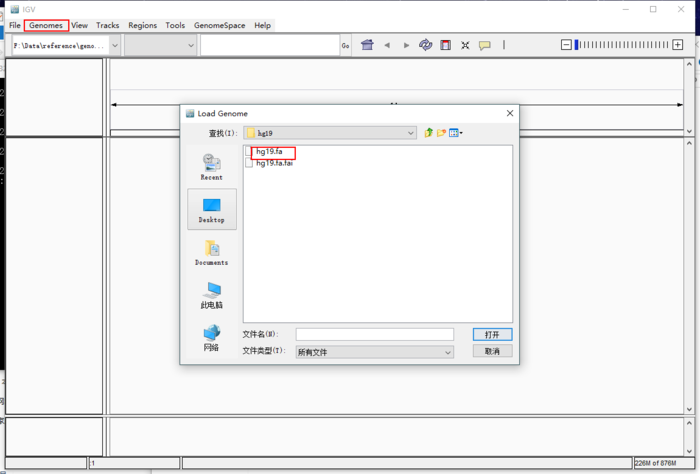
通过genome -> Load Genome From Files加载之前得到基因组文件。
进一步,还需要加载gff基因注释文件,File -> Load From Files
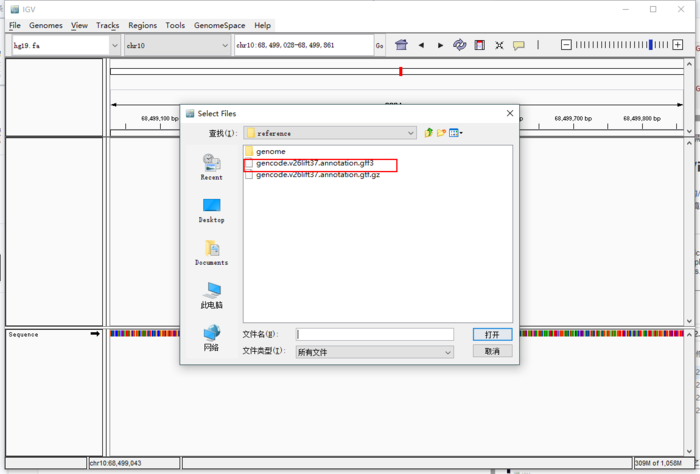
显示未排序出错,可以使用Tool -> Run igvtools,进行排序。
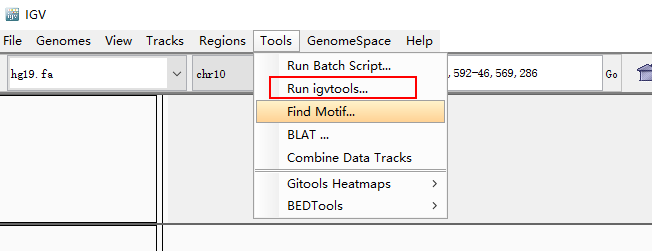
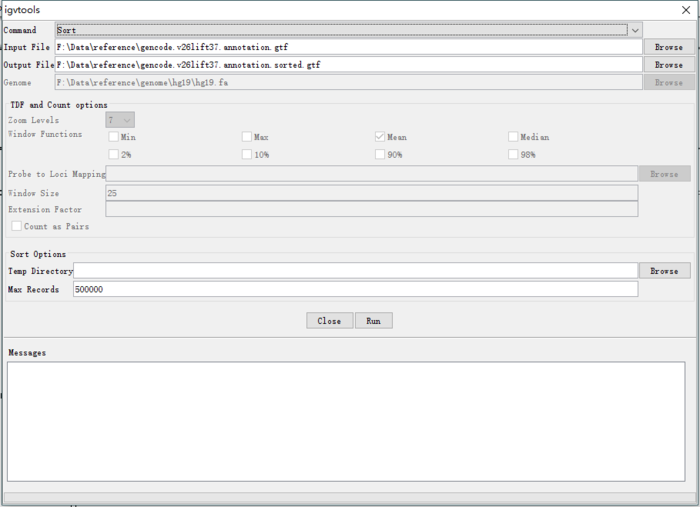
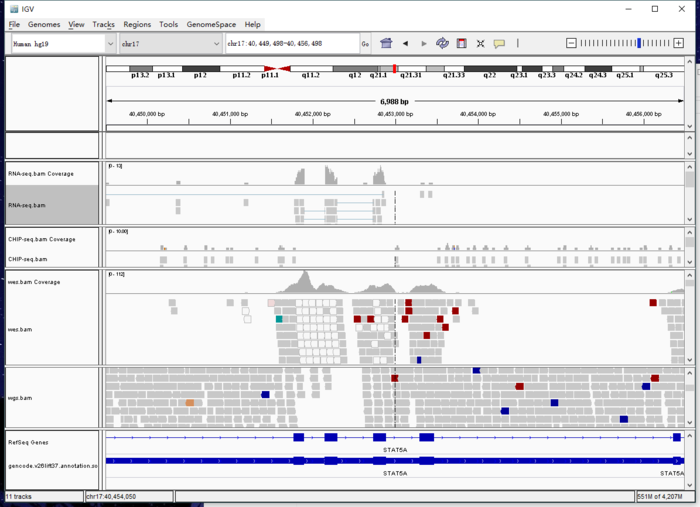
之后就可以重新加载排序后的gtf文件进行操作。生信宝典写过一篇文章介绍测序数据可视化(http://mp.weixin.qq.com/s/Q7pqycmQH58xU6hw_LECWA) 我也在看文档摸索中,先放上基因截图
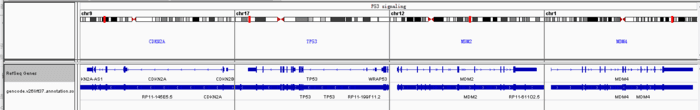
下面这张图是对高通量测序的理解,提供数据的截图
1F
转录组入门(4):了解参考基因组及基因注释