

染色质免疫共沉淀测序(ChIP-Seq)是指对染色质免疫共沉淀(ChIP)获得的DNA片段进行大规模测序,并能把所研究蛋白的DNA结合位点精确定位到基因组上。
Roche GS FLX Titanium 、Illumina Solexa GA IIx和AB SOLID 4 这3种测序技术均可以用于ChIP-seq,其中采用Illumina Solexa GA IIx进行ChIP-Seq已有较多文献报道。
ChIP-Seq技术高质量、高通量、低成本的数据产出,为表观遗传组学研究奠定了技术基础。研究者可以在以下几方面展开研究:(1)判断DNA链的某一特定位置会出现何种组蛋白修饰;(2)检测RNA polymerase II及其它反式因子在基因组上结合位点的精确定位;(3)研究组蛋白共价修饰与基因表达的关系;(4)CTCF转录因子研究。
ChIP-Seq有什么样品要求?
答:(1) 请提供浓度≥10 ng/ul、总量≥200 ng、OD260/280为1.8~2.2的DNA样品;若单次ChIP后DNA量不够,建议将2~3次ChIP的DNA合并在一起。
(2) 请提供DNA打断时检测胶图,要求打断后DNA电泳主带在200-500 bp范围内;请对于ChIP获得 DNA设计引物进行QPCR验证和定量,能够提供检测位点的检测报告。附阳性和阴性对照。
(3) 样品请置于1.5 ml管中,管上注明样品名称、浓度以及制备时间,管口使用Parafilm封口。在运输前将所有样品管固定于50 ml带盖离心管中,再将50 ml管放在封口袋中。为了防止低浓度样品黏附在离心管壁上,请使用non-stick tube运输DNA ;建议使用冰袋运输,并且尽量选用较快的邮递方式,以降低运输过程中样品降解的可能性。
ChIP-Seq相比ChIP-chip有哪些优势?
答:第一,ChIP-Seq 能实现真正的全基因组分析。目前所能获得的芯片上固定的探针只能代表全基因组部分序列,所获得的杂交信息具有偏向性;第二,对于结合位点分析,ChIP-Seq 通过寻找“峰”,结合分辨率可精确到10~30 bp,而芯片上探针由于长度所限,无法精确定位,即使目前最高水平的商业芯片都无法提供可与ChIP-Seq 媲美的分辨率;第三是所需样本数量。ChIP-chip 需要多达4~5 μg 的起始样本,在杂交之前需要进行LM-PCR,但可能导致背景增高,竞争性扩增等导致假阳性。而ChIP-Seq 仅需要纳克级起始材料,如SOLiD 起始材料可低至20ng。两者技术特点如下:
研究方法 | CHIP-on-chip | CHIP-Seq |
| 分辨率 | 30~100bp | 1bp |
| 覆盖范围 | 受芯片容量限制,只能选择性地扫描特定区域,无法覆盖全基因组 | 只要测定的序列(Reads)能够定位到基因组上,就能获得全部基因组信息 |
| 缺陷 | 探针和非特异性区域杂交 | 测序数据会有一些GC含量偏向 |
| 性价比 | 只能研究在基因组上广泛存在的目的位点(Broading bingding) | 可以扫描全基因组;可以研究在基因组上存在的稀有目的位点(Sharp bingding) |
| 需要的DNA量 | 高 | 低(10~50bp) |
| 动态量程 | 弱信号会被遗弃;强信号会饱和 | 没有局限 |
| 选择数据产出量 | 不可以 | 可以 |
ChIP-Seq测序文库的构建方法?
答:目前为止,唯有Solexa在ChIP-seq领域被广泛报道,采用Solexa测序其ChIP-seq测序文库构建方法如下
(1)ChIP富集DNA片段(图1)。首先用甲醛将活体细胞交联,随后将细胞核内的染色质用超声波打断成目标长度(通常0.2-1kb)的短片段形式,用所研究的蛋白特异性抗体将蛋白结合的DNA片段免疫共沉淀,接下来将蛋白质-DNA复合物解交联并纯化DNA片段。
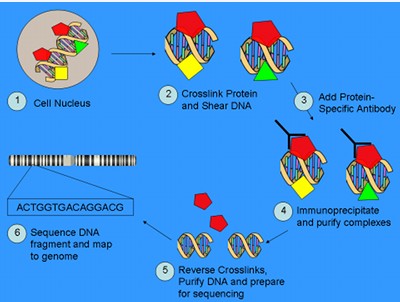 ChIP富集DNA片段示意图
ChIP富集DNA片段示意图
(2)构建测序文库。对于ChIP富集的DNA片段纯化后,经末端补平,3’末端加A,连接接头一系列处理后,进行电泳割胶,回收150-300bp范围左右的片段,然后对回收的DNA片段进行PCR扩增,上机测序。所测序列首先进行基因组比对然后用不同的算法进行结合位点的统计。
样品制备过程中是否需要PCR扩增?PCR扩增后是否会影响最后的结果?还有那些因素会影响ChIP-Seq的结果?
答:由于ChIP下来的DNA样品量非常少,所以在样品制备过程中需要经过一步PCR扩增的过程,PCR扩增主要是为了获得足够上机反应的DNA量。如果客户能够提供足够量的DNA样品,我们不再进行PCR扩增。由于是线性扩增,扩增前后的结果很相似,基本上不会影响测序结果。抗体的质量,特异性,实验设计,ChIP的实验操作,DNA片段长度范围,测序通量,测序质量等都会影响ChIP-Seq的结果。
ChIP-Seq测序对照的选择?
答:ChIP-seq过程中,由于DNA富集过程受多种因素的影响。因此,在做ChIP实验时,一定要做好实验对照。因为没有对照,很难对实验结果的可靠性进行评估。一般有三种实验对照:Input对照、阳性对照和阴性对照。
(1)Input对照:在进行免疫沉淀前,需要取一部分断裂后的染色质做Input对照。Input是断裂后的基因组DNA,需要与沉淀后的样品DNA一起经过逆转交联,DNA纯化,以及最后的PCR或其他方法检测。Input对照不仅可以验证染色质断裂的效果,还可以根据Input中的靶序列的含量以及染色质沉淀中的靶序列的含量,按照取样比例换算出ChIP的效率,所以Input对照是ChIP实验必不可少的步骤。
(2)阳性对照和阴性对照:阳性抗体和阴性抗体对照是最基本的实验对照。阳性抗体通常选择与已知序列相结合的比较保守的蛋白的抗体,常用的包括组蛋白抗体或RNA Polymerase II抗体等。阴性抗体通常选择目的蛋白抗体宿主的IgG或血清。目的蛋白抗体的结果与阳性抗体和阴性抗体的结果相比较,才能得出正确结论。如果目的蛋白没有商品化的适用于染色质免疫沉淀实验的抗体,只有其他用途的抗体时,可以先做蛋白质免疫沉淀(Immunoprecipitation)检测。如果抗体可以成功的沉淀蛋白,再进行染色质免疫沉淀实验的检测。
怎样进行ChIP-seq多重测序(Multiplexed Sequencing)?
答:根据所研究的ChIP-DNA的大小以及所选择的测序深度,计算每个样品所需的数据通量,在单个个体的数据量能够保证的前提下,可以对多个样品进行多重测序(Multiplexed Sequencing)。Solexa 运行1个run最多可以获取50Gb的碱基数据,共8个通道,每个通道可以对12个混合样本进行测序,每个run可以对96个混合样本进行测序;多重测序(Multiplexed Sequencing)的步骤如下:(1)对富集得到的双链DNA进行末端修复;(2)将“A”碱基加入到DNA片段的3’末端;(3)使用特定的测序接头连接DNA片段两端;(4)纯化连接产物以除去未连接的接头序列;(5)以Barcodes标记的引物和高保真聚合酶PCR扩增的DNA片段;(6)检测测序文库。
只要采用高通量测序,就一定可以达到高分辨率?导致ChIP-seq测序假阳性比较高的因素?
答:高通量的测序确实大大提高了ChIP检测的分辨率,但并不是高分辨率的唯一决定因素。免疫富集后,染色质被打断的片段长度也会影响分辨率。DNA打碎方法、染色质开放程度的不均一性、PCR扩增偏向性、基因组的重复程度以及测序和序列比对过程中的错误都会引入系统误差造成假阳性,测序后首先将序列比对到已知基因组上并确立真正的结合位点(峰,peak)。对于转录因子,要寻找“峰”对应的下游调控基因(靶基因),或者构建转录因子结合位点的保守结合序列,如果转录因子的motif是已知的,则可以计算“峰”序列中包含motif序列的百分比,间接估计实验结果的可靠性。