

ATAC-Seq/ChIP-Seq通常需要2个会更多的生物学重复(十分珍贵或者稀有样本除外,但必须做至少2次技术重复)。理论上重复样本的peaks应该有高度的一致性,实际情况并不完全与预期一致。如何评价重复样本的重复性的好坏?如何得到一致性的peaks?这里介绍两种方法:
1. 用Bedtools进行简单的overlap合并重复样本
2. IDR(Irreproducibility Discovery Rate)的方法获得高重复性的peaks
1. Overlapping peaks using bedtools
如何得到两个重复样本间一致性的peaks? 一种简单粗暴的方法就是用bedtools计算peaks的overlaps。
用法:bedtools intersect [OPTIONS] -a <bed/gff/vcf/bam> -b <bed/gff/vcf/bam>
-a: 参数后加重复样本1(A)
-b:参数后加重复样本2(B),也可以加多个样本
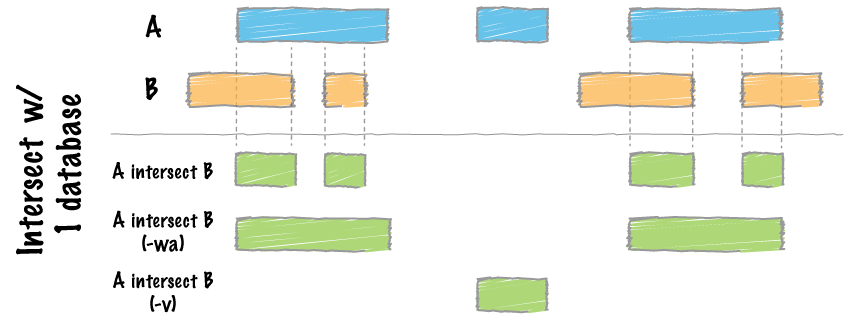
其他常用参数解释和图解如下:
-wo:Write the original A and B entries plus the number of base pairs of overlap between the two features.
-wa:Write the original entry in A for each overlap.
-v:Only report those entries in A that have no overlaps with B
如果只有-a和-b参数,返回的是相对于A的overlaps。加上参数-wo返回A和B原始的记录加上overlap的记录。参数-wa返回每个overlap中A的原始记录。
使用代码示例:
bedtools intersect \ -a macs2/Nanog-rep1_peaks.narrowPeak \ -b macs2/Nanog-rep2_peaks.narrowPeak \ -wo > bedtools/Nanog-overlaps.bed
2. Irreproducibility Discovery Rate (IDR)
评估重复样本间peaks一致性的另一种方法是IDR。IDR是通过比较一对经过排序的regions/peaks 的列表,然后计算反映其重复性的值。
IDR在ENCODE和modENCODE项目中被广泛使用,也是ChIP-seq指南和标准中的一部分。
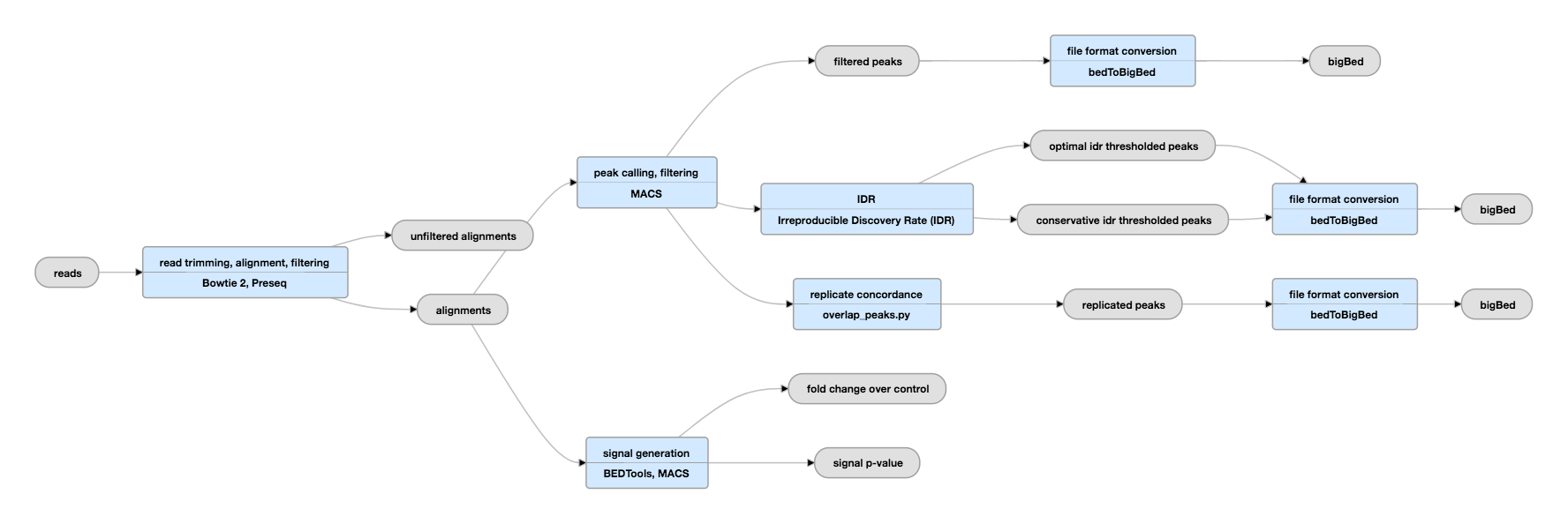
IDR的 优点:
- 避免了初始阈值的选择,解决了不同callers的不可比较性
- IDR不依赖于阈值的选择,所有regions/peaks都被考虑在内。
- 它是依赖regions/peaks的排序,不要求对输入信号进行校准或标准化
IDR的详细说明参考: - https://github.com/nboley/idr
- https://github.com/hbctraining/In-depth-NGS-Data-Analysis-Course/blob/master/sessionV/lessons/06_handling-replicates.md#irreproducibility-discovery-rate-idr
使用IDR的注意事项:
- 建议使用IDR时,MACS2 call peaks的步骤参数设置不要过于严格,以便鉴定出更多的peaks。
- 使用IDR需要先对MACS2的结果文件narrowPeak根据
-log10(p-value)进行排序。
# Call peaks macs2 callpeak -t sample.final.bam -n sample --shift -100 --extsize 200 --nomodel -B --SPMR -g hs --outdir Macs2_out 2> sample.macs2.log #Sort peak by -log10(p-value) sort -k8,8nr NAME_OF_INPUT_peaks.narrowPeak > macs/NAME_FOR_OUPUT_peaks.narrowPeak
- 使用IDR示例
idr --samples sample_Rep1_sorted_peaks.narrowPeak sample_Rep2_sorted_peaks.narrowPeak \ --input-file-type narrowPeak \ --rank p.value \ --output-file sample-idr \ --plot \ --log-output-file sample.idr.log
--samples:narrowPeak的输入文件(重复样本)
--input-file-type:输入文件格式包括narrowPeak,broadPeak,bed
--rank p.value:以p-value排序
--output-file: 输出文件路径
--plot:输出IDR度量值的结果
输出文件解读:
详细内容可参考:https://github.com/nboley/idr#output-file-format
输出文件包括:
- sample-idr
- sample-idr.log
- sample-idr.png
(1)sample-idr
sample-idr是common peaks的结果输出文件,格式与输入文件格式类似,只是多了几列信息。前10列是标准的narrowPeak格式文件,包含重复样本整合后的peaks信息。
- 第5列:包含缩放的 IDR 值
- score int
如min(int(log2(-125IDR), 1000),那么IDR=0,缩放的IDR就是1000;IDR=0.05, int(-125log2(0.05)) = 540;IDR=1.0, int(-125log2(1.0) = 0。
- score int
- 第11列和第12列:分别是local和global IDR值
- col11: localIDR float -log10(Local IDR value)
- col12: globalIDR float -log10(Global IDR value)
global IDR值是第5列中用于计算缩放的IDR值,类似于对p值进行多个假设校正以计算FDR;local IDR类似于属于不可重复噪声部分的峰值的后验概率。详细内容可阅读Measuring reproducibility of high-throughput experiments。
- 第13、14、15、16列:是重复样本1相关的信息。
- col13: rep1_chromStart int
- col14: rep1_chromEnd int
- col15: rep1_signalValue float
- col16: rep1_summit int
- 第17-20列:是重复样本2相关的信息,和13-16四列信息类似。
其他列信息如下:
- col1: chrom string
- col2: chromStart int
- col3: chromEnd int
- col4: name string
- col6: strand [+-.] Use '.' if no strand is assigned.
- col7: signalValue float
- col8: p-value float
- col9: q-value float
- col10: summit int
wc -l *-idr 计算下common peaks的个数,接着可再计算下与总peaks的比率。
如果想看IDR<0.05的,可以通过第5列信息过滤:
awk '{if($5 >= 540) print $0}' sample-idr | wc -l
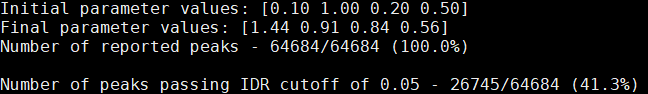
(2)sample-idr.log
log文件会给出peaks通过IDR < 0.05的比率,如下图所示
3)sample-idr.png
png文件包括4个图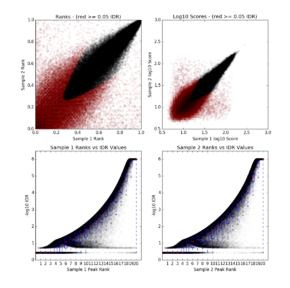
左上: Rep1 peak ranks vs Rep2 peak ranks, 没有通过特定IDR阈值的peaks显示为红色。
右上:Rep1 log10 peak scores vs Rep2 log10 peak scores,没有通过特定IDR阈值的peaks显示为红色。
下面两个图: Peak rank vs IDR scores,箱线图展示了IDR值的分布,默认情况下,IDR值的阈值为-1E-6。
参考资料: