

ATAC-Seq分析教程系列
ATAC-Seq分析教程:ATAC-seq的背景介绍以及与ChIP-Seq的异同
ATAC-Seq分析教程:用MACS2软件call peaks
ATAC-Seq分析教程:对ATAC-Seq/ChIP-seq的质量评估(一)phantompeakqualtools
ATAC-Seq分析教程:对ATAC-Seq/ChIP-seq的质量评估(二)ChIPQC
ATAC-Seq分析教程:用ChIPseeker对peaks进行注释和可视化
ATAC-Seq分析教程:用网页版工具做功能分析和motif分析
学习目标
- 学习用DiffBind流程评估两个样本间的差异结合区域
- 用PCA评估样本间的关系
- 评估不同工具得到的差异peaks的一致性
评估差异peaks富集的工具
ATAC-Seq下游分析的另一个重点是差异peaks分析。如分析不同的实验条件、多个时间节点、不同的发育时期等的差异区域。鉴定这些差异peaks区域在生物医学研究中也具有重要意义,目前也有多种相关的工具被开发:
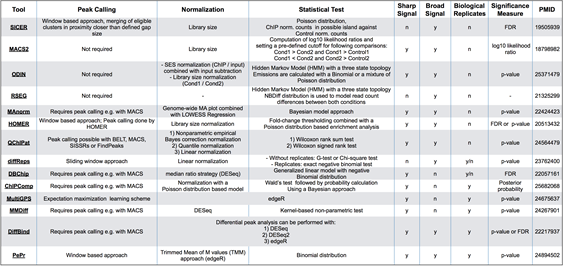
选择合适的工具需考虑以下几个因素:
- 所用的软件不需要大量的代码移植,该工具是否被维护和频繁更新。
- 用户需要提供什么样的输入文件,是否与peaks calling所用工具可以衔接。
- 信号分布的基础统计模型是什么?是基于泊松分布或更灵活的负二项分布。
- 一些工具是专门针对特定的ATAC-seq或ChIP-seq 数据(信号类型)设计的,如组蛋白修饰或转录因子(TF)结合。
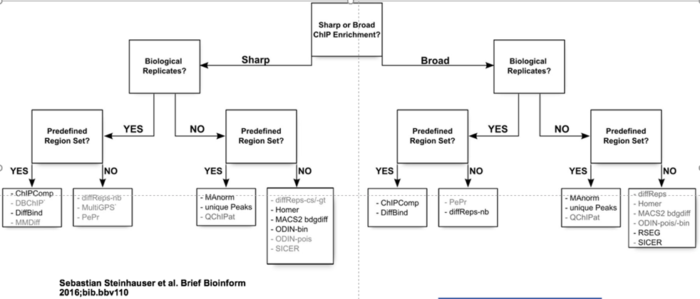
具体选择哪种工具决定于实验设计,下面的决策树可以帮助缩小你的选择范围。
DiffBind
DiffBind是鉴定两个样本间差异结合位点的一个R包。主要用于peak数据集,包括对peaks的重叠和合并的处理,计算peaks重复间隔的测序reads数,并基于结合亲和力鉴定具有统计显著性的差异结合位点。适用的统计模型有DESeq、DESeq2、edgeR。详细内容可参考DiffBind的文档:http://bioconductor.org/packages/release/bioc/vignettes/DiffBind/inst/doc/DiffBind.pdf
使用方法:
1. 下载安装DiffBind
source("https://bioconductor.org/biocLite.R")
biocLite("DiffBind")
# view documentation
browseVignettes("DiffBind")2. 准备输入文件
需要准备一个SampleSheet文件,与ChIPQC的方法一样。SampleSheet文件是根据实验设计和数据存储地址等信息创建的一个csv格式文件,包含的表头信息有“SampleID”、 “Tissue”、 “Factor”、 “Condition” 、”Treatment”、”Replicate” 、”bamReads” 、”ControlID”、 “bamControl” “Peaks”、 “PeakCaller”(bam,peak文件分别在比对和call peak的步骤产生)。
![]()
3. 差异peaks分析
读入文件
一旦读入了peaksets,合并函数就找到所有重叠的peaks,并导出一致性的peaksets。
library(DiffBind) dbObj <- dba(sampleSheet="SampleSheet.csv")
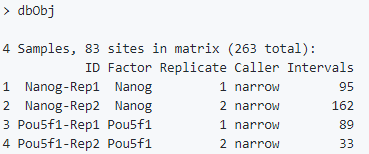
亲和结合矩阵
计算每个peaks/regions的count信息。先对一致性的peaks数据集进行标准化,然后根据他们的峰值(point of greatest read overlap)再次中心化并修剪一些peaks,最终得到更加标准的peak间隔。使用函数dba.count(),参数bUseSummarizeOverlaps可以得到更加标准的计算流程。
dbObj <- dba.count(dbObj, bUseSummarizeOverlaps=TRUE)
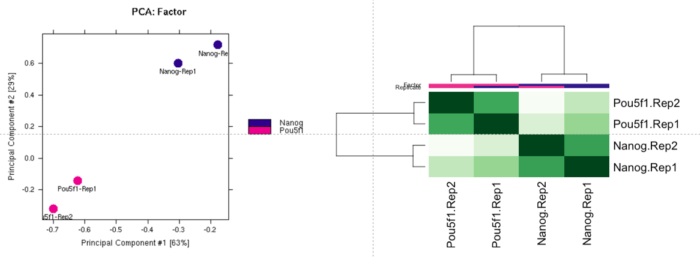
dba.plotPCA(dbObj, attributes=DBA_FACTOR, label=DBA_ID)
plot(dbObj)结果中同时计算了FRiP (质量评估的一个标准,可以参考对ATAC-Seq/ChIP-seq的质量评估)。
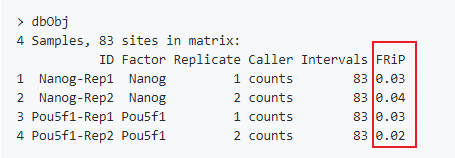
样本间的聚类:
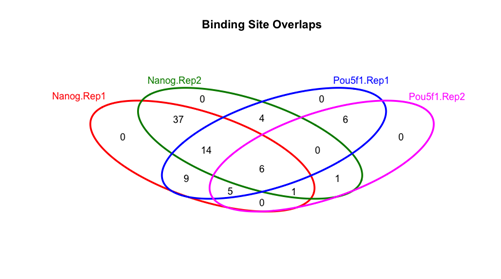
Venn图展示样本间peaks的重合
差异分析
差异分析是DiffBind的核心功能,默认情况是基于DEseq2, 可以设置参数method=DBA_EDGER选择edgeR,或者设置method=DBA_ALL_METHODS。每种方法都会评估差异结果的p-vaue和FDR。
# Establishing a contrast
dbObj <- dba.contrast(dbObj, categories=DBA_FACTOR,minMembers = 2)
dbObj <- dba.analyze(dbObj, method=DBA_ALL_METHODS)
# summary of results
dba.show(dbObj, bContrasts=T)
# overlapping peaks identified by the two different tools (DESeq2 and edgeR)
dba.plotVenn(dbObj,contrast=1,method=DBA_ALL_METHODS)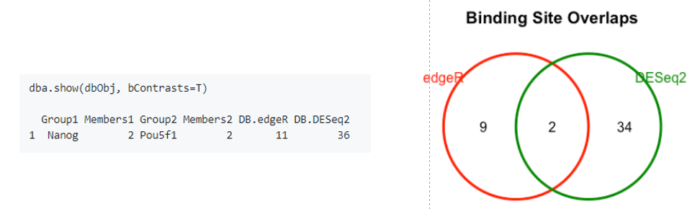
提取结果
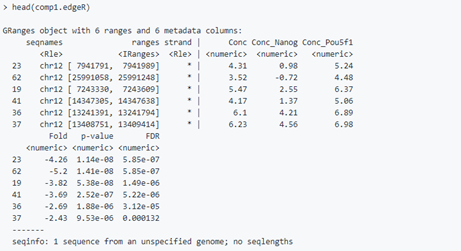
comp1.edgeR <- dba.report(dbObj, method=DBA_EDGER, contrast = 1, th=1)
comp1.deseq <- dba.report(dbObj, method=DBA_DESEQ2, contrast = 1, th=1)结果文件包含所有位点的基因组坐标,以及差异富集的统计数据包括fold change、p值和FDR。
其中Conc的表示read的平均浓度,即对peak 的read counts进行log2标准化
The value columns show the mean read concentration over all the samples (the default calculation uses log2 normalized ChIP read counts with control read counts subtracted) and the mean concentration over the first (Resistant) group and second (Responsive) group.
保存文件
# EdgeR
out <- as.data.frame(comp1.edgeR)
write.table(out, file="results/Nanog_vs_Pou5f1_edgeR.txt", sep="t", quote=F, col.names = NA)
# DESeq2
out <- as.data.frame(comp1.deseq)
write.table(out, file="results/Nanog_vs_Pou5f1_deseq2.txt", sep="t", quote=F, col.names = NA)以bed格式保存显著性的差异结果
# Create bed files for each keeping only significant peaks (p < 0.05)
# EdgeR
out <- as.data.frame(comp1.edgeR)
edge.bed <- out[ which(out$FDR < 0.05),
c("seqnames", "start", "end", "strand", "Fold")]
write.table(edge.bed, file="results/Nanog_vs_Pou5f1_edgeR_sig.bed", sep="t", quote=F, row.names=F, col.names=F)
# DESeq2
out <- as.data.frame(comp1.deseq)
deseq.bed <- out[ which(out$FDR < 0.05),
c("seqnames", "start", "end", "strand", "Fold")]
write.table(deseq.bed, file="results/Nanog_vs_Pou5f1_deseq2_sig.bed", sep="t", quote=F, row.names=F, col.names=F)