

一、基础知识
1. 解密表观遗传学的三个方向与测序方法
- 1. 探索染色质的开放性(chromatin accessibility)
ATAC-seq: Assay of Transposase Accessible Chromatin sequencing
DNase-seq: DNase I hypersensitive sites sequencing
FAIRE-seq: Formaldehyde-Assisted Isolation of Regulatory Elements sequencing - 2. 探索转录因子的绑定与组蛋白修饰(TF binding and histone modifications)
ChIP-seq: Chromatin Immuno-Precipitation sequencing - 3. 探索核小体占位(nucleosome positioning and occupancy)
MNase-seq: Micrococcal Nuclease sequencing
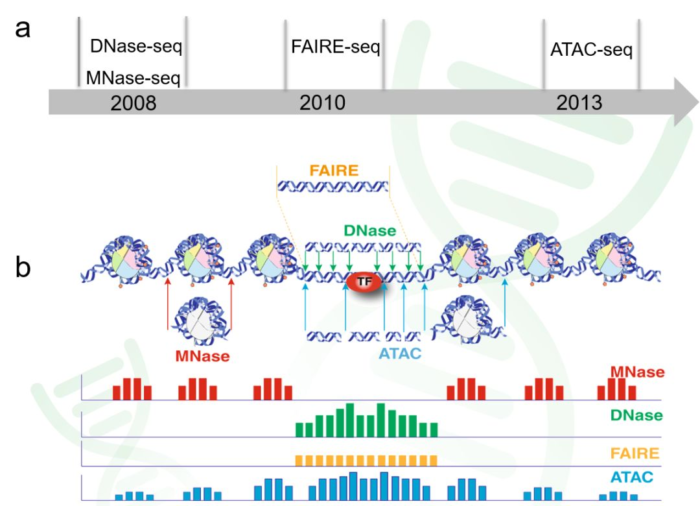
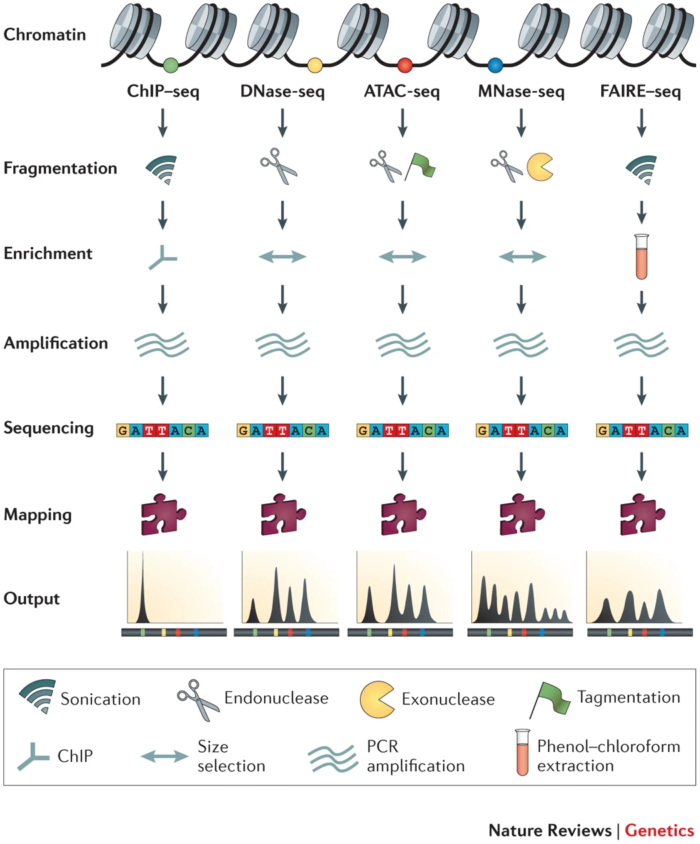
2. 名词解释
epigenetics:表观遗传学。表观遗传学修饰在不改变DNA序列的情况下控制着基因的表达,包括染色质重塑、组蛋白修饰、DNA甲基化和microRNA通路等peaks: 峰。常用来表示染色质的开放程度,因为是测序的reads落在了染色质的开放区,堆叠后被可视化的一种丰度的体现。
但peaks到底代表什么?莫非peaks所在区域要么是启动子要么是增强子?再或者就是笼统定义为调控元件?(我目前的观点是,只能把他理解为酶切的位点,如果非要定义是否是特定分调控元件,只能进行后续分析。)
THSs: 转座酶超敏感位点(transposase hypersensitive sites)。CREs: 顺式调控元件(cis-regulatory elements)。即DNA分子中具有转录调节功能的特异DNA序列。按功能特性,真核基因顺式作用元件分为启动子、增强子及沉默子。ACRs: 染色质开放区域(accessible chromatin regions)。即正常或核小体被酶切裸露出来的DNA片段所在的区域。这篇【NP | 2019】根据ACRs距离最近基因的距离将ACRs分为三种类型:genic (gACRs; overlapping a gene), proximal (pACRs; within 2kb of a gene) or distal (dACRs; >2 kb from a gene),分别是跨越基因的,近端的,远端的染色质开放区。transposon: 转座子。一段可以从原位上单独复制或断裂下来,环化后插入另一位点,并对其后的基因起调控作用的DNA序列。promoter: 启动子。启动子是位于结构基因5'端上游的DNA序列,能活化RNA聚合酶,使之与模板DNA准确的结合并具有转录起始的特异性。每个启动子包括至少一个转录起始点以及一个以上的功能组件(典型的如TATA盒子)proximal promoters: 近端启动子。是DNA上位于基因开始之前的一个区域,在那里蛋白质和其他分子结合在一起准备读取该基因。enhancer: 增强子。增强子是远离转录起始点、决定基因的时间、空间特异性表达、增强启动子转录活性的DNA序列,其发挥作用的方式通常与方向、距离无关,可位于转录起始点的上游或下游。从功能上讲,没有增强子存在,启动子通常不能表现活性;没有启动子时,增强子也无法发挥作用。根据南京大学陈迪俊老师的研究表明增强子比启动子能结合更多的转录因子(Nature Communications)TFs: 转录因子(transcription factors)是保证目的基因以特定的强度在特定的时间与空间表达的蛋白质分子。与RNA聚合酶Ⅱ形成转录起始复合体,共同参与转录起始的过程。TSS: 转录起始位点(transcription start site)。在一个典型的基因内部,排列顺序为转录起始位点(TSS,一个碱基)-起始密码子编码序列 (ATG)-终止密码子编码序列(TGA)-转录终止位点 (TTS),即TSS-ATG-TGA-TTShistone:组蛋白。通常含有H1,H2A,H2B,H3,H4等5种成分,其中H1与H3极度富含赖氨酸(lysine),H1不保守,其他组蛋白的基因非常保守。除H1外,其他4种组蛋白均分别以二聚体(共八聚体)相结合,形成核小体核心。DNA便缠绕在核小体的核心上。而H1则与核小体间的DNA结合nucleosome: 核小体。是由DNA和组蛋白形成的染色质基本结构单位。每个核小体由146bp的DNA缠绕组蛋白八聚体1.75圈形成。核小体核心颗粒之间通过50bp左右的连接DNA相连,暴露在核小体表面的DNA能被特定的核酸酶接近并切割H3K4me1: 组蛋白H3上的第4位赖氨酸发生单甲基化(mono-methylation of H3 at lysine 4) (NG | H3K4me1与增强子的关系)H3K9ac:组蛋白H3上的第9位赖氨酸发生乙酰化(H3 acetylation marks at lysine 9)关于H3K4me1与H3K9ac的不同,依旧可以参考这篇文献【NP | 2019】或【NC | 2019】组蛋白甲基化:甲基化可发生在组蛋白的赖氨酸和精氨酸残基上,而且赖氨酸残基能够发生单、双、三甲基化,而精氨酸残基能够单、双甲基化,这些不同程度的甲基化极大地增加了组蛋白修饰和调节基因表达的复杂性组蛋白乙酰化:四种类型的组蛋白相互作用,将细胞核里的DNA紧紧地包装起来。这样的紧密包装能够有效阻止酶读取DNA上的遗传信息。然而,乙酰基连到组蛋白上能削弱它们对DNA的占据。因此局部乙酰化能暴露出相应的基因,让它们更容易激活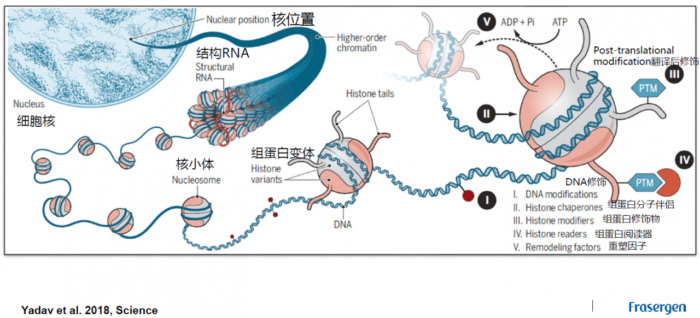 真核生物细胞核内染色质结构
真核生物细胞核内染色质结构
二、基础分析
1. 分析前的准备
1.1 下载ATAC测序数据(或者用你自己的数据也行)
- 所需软件:Aspera Connect
- 软件安装与使用:参考这系列文章
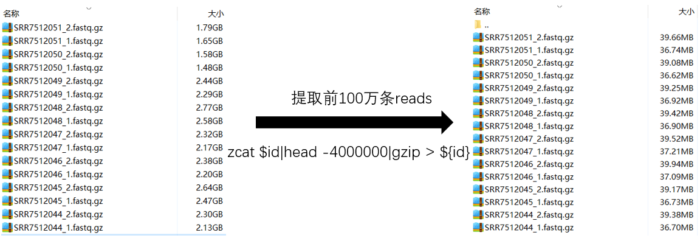
- 数据下载:项目ID【PRJNA480717】【文章原文】,SRR序列号如下图,下载方法和上文相同。由于数据量大,测试浪费时间,所以我在每个文件中取了100w条reads,由于百度网盘太慢了,你可以去扣扣群559758504里面随便下载一个用于测试。
# 提取100w条reads代码
ls *gz|while read id;do zcat $id|head -4000000|gzip > ./100/${id};done
1.2 下载tair10基因组与gff3注释文件
wget https://www.arabidopsis.org/download_files/Genes/TAIR10_genome_release/TAIR10_chromosome_files/TAIR10_chr_all.fas wget https://www.arabidopsis.org/download_files/Genes/TAIR10_genome_release/TAIR10_gff3/TAIR10_GFF3_genes_transposons.gff # 改名 mv TAIR10_chr_all.fas TAIR10_genome.fa mv TAIR10_GFF3_genes_transposons.gff TAIR10.gff3
1.3 安装miniconda
- 由于后续分析大多数软件都是来源于conda,因此你必须知道怎么使用。参考【conda的安装与使用】
1.4 所需软件安装(分析中所用到的软件都汇集于此,下文不再介绍如何下载)
请先激活conda环境在安装!
conda install -c bioconda -y fastqc conda install -c bioconda -y trimmomatic conda install -c bioconda -y bwa conda install -c bioconda -y samtools conda install -c bioconda -y picard conda install -c bioconda -y macs2 conda install -c bioconda -y bedtools conda install -c bioconda -y deeptools conda install -c bioconda -y homer
后续分析是使用SRR7512044作为测试数据,但是思路和方法都可以借鉴。
2. 测序质量检测
fastqc -t 8 -o ./ *.fastq.gz
-t: 线程
-o: 存放路径,不用指定前缀,默认为.fastq.gz前面的字段
*.fastq.gz: fastqc可以同时接受多个fastq.gz文件,因此采用正则表达式【*】表示全部
- 结果解读
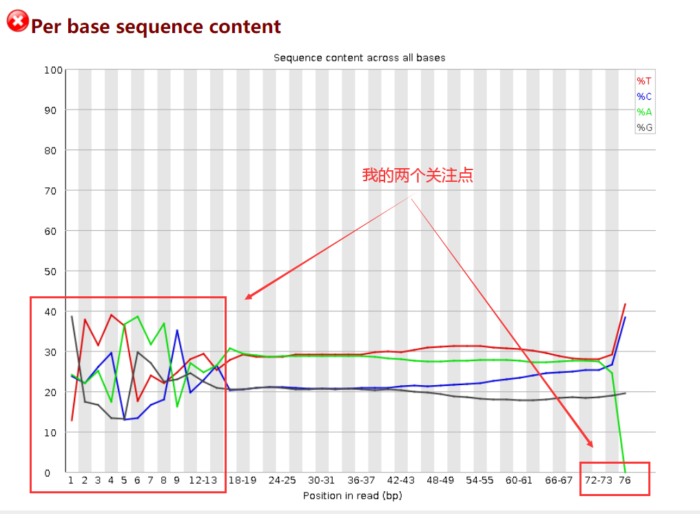
由于开始测序时不准确,因此前15bp左右的ATGC含量有波动,需要在后面截掉。而最长reads为76bp,则暗示着我最后至少要保留多长的序列。
一般我的计算思路是:保留的序列长度 =(最长的的reads长度-前面波动的碱基长度)× 40%
2. 测序质量检控制
- 所需软件:Trimmomatic【官方网站】【中文介绍】
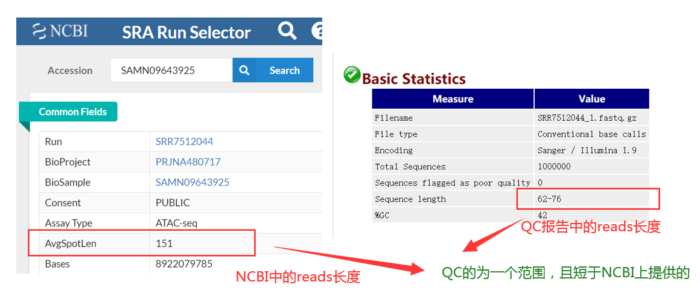
其实这批数据已经做过质控了,如何判断的呢?QC的为一个范围且短于提供给NCBI上的数据。因此我们没必要质控,但为了流程的完整性,还是假装质控一下。质控之后,还要进行质检,如此循环,直到合格。
# 定义一些变量,后面修改起来方便
filename=SRR7512044
trimmomatic PE -threads 10
./${filename}_1.fastq.gz ./${filename}_2.fastq.gz
./${filename}_1.clean.fq.gz ./${filename}_1.drop.fq.gz
./${filename}_2.clean.fq.gz ./${filename}_2.drop.fq.gz
HEADCROP:15 LEADING:3 TRAILING:3 SLIDINGWINDOW:4:15 MINLEN:24
PE: 双端模式。需要两个输入文件,正向测序序列和反向测序序列:sample_R1.fastq.gz和sample_R2.fastq.gz;
以及四个输出文件:sample_1.clean.fq.gz和sample_1.drop.fq.gz;sample_2.clean.fq.gz和sample_2.drop.fq.gz
-threads: 线程数
HEADCROP: 从 reads 的开头切掉指定数量的碱基,即从fastqc中看的
LEADING: 从 reads 的开头切除质量值低于阈值的碱基
TRAILING: 从 reads 的末尾开始切除质量值低于阈值的碱基
SLIDINGWINDOW: 从 reads 的 5' 端开始,进行滑窗质量过滤,切掉碱基质量平均值低于阈值的滑窗
MINLEN: 如果经过剪切后 reads 的长度低于阈值则丢弃这条 reads,即上面的计算方法:(76-15)×40%≈24
3. 建立索引与序列比对
- 所需软件:bwa和samtools
- 建立基因组索引。需要的时间挺长的,建议放后台。建好之后以后就可以跳过这一步了,且在经典的比对软件STAR、hisat2、bowtie2和bwa-mem中我更喜欢使用STAR和bwa-mem。本次使用bwa-mem操作演示
bwa index -p /路径/bwaIndexTair /路径/TAIR10_genome.fa
-p: 索引前缀,后缀自动补充
TAIR10_genome.fa: 基因组文件
- 序列比对
由于sam文件较大,因此我们直接跳过sam,使用samtools转换为排序后的bam文件,注意【|】不要漏掉,且排序是依据reads比对到基因组的位置排的
# 定义一些变量,后面用
filename=SRR7512044
index=/路径/bwaIndexTair
bwa mem -M -t 8
-R "@RGtID:${filename}tSM:${filename}tLB:WXStPL:Illumina"
$index
${filename}_1.clean.fq.gz
${filename}_2.clean.fq.gz
| samtools sort -O bam -@ 10 -o ./${filename}.raw.bam
#无论干啥,生成bam文件后就要建立bam索引,并检查比对情况
samtools index -@ 10 -b ./${filename}.raw.bam
samtools flagstat ./${filename}.raw.bam > ./${filename}.raw.stat
cat ${filename}.raw.stat
-M: 将 shorter split hits 标记为次优,以兼容 Picard’s markDuplicates 软件
-R: STR 完整的read group的头部,可以用 't' 作为分隔符, 在输出的SAM文件中被解释为制表符TAB. read group 的ID,会被添加到输出文件的每一个read的头部,
-O: 表示输出的bam文件
-o: 输出的bam文件名
-t与@: 线程数
- 关于stat中内容的解释如下
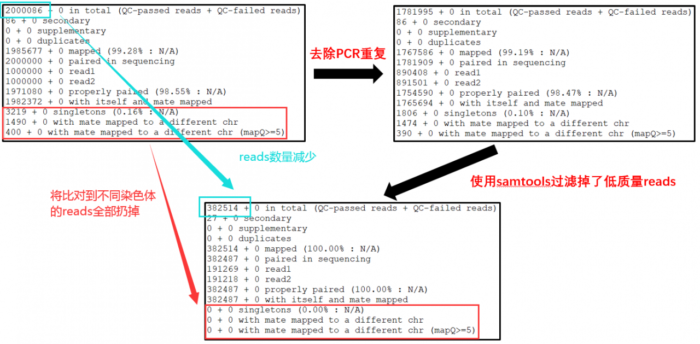
14608455 0 in total (QC-passed reads QC-failed reads) reads总数 37967 0 secondary 出现比对到参考基因组多个位置的reads数 0 0 supplementary 可能存在嵌合的reads数 0 0 duplicates 重复的reads数 14590894 0 mapped (99.88% : N/A) 比对到参考基因组上的reads数 14570488 0 paired in sequencing 属于PE read的reads总数。 7285244 0 read1 PE read中Read 1 的reads 总数。 7285244 0 read2 PE read中Read 2 的reads 总数。 14507068 0 properly paired (99.56% : N/A) 完美比对的reads总数。PE两端reads比对到同一条序列,且根据比对结果推断的插入片段大小符合设置的阈值。 14551500 0 with itself and mate mapped PE两端reads都比对上参考序列的reads总数。 1427 0 singletons (0.01% : N/A) PE两端reads,其中一端比上,另一端没比上的reads总数。 26260 0 with mate mapped to a different chr PE read中,两端分别比对到两条不同的序列的reads总数。 17346 0 with mate mapped to a different chr (mapQ>=5) PE read中,两端分别比对到两条不同
4. 去除PCR重复并再次进行质控
- 所需软件:picard与samtools
不建议使用sambamba,虽然快,但是毛病多,尤其是在你不知情的情况下,软件卡死或者在最后输出文件时提示输出文件夹过多等。
picard MarkDuplicates
REMOVE_DUPLICATES=true
I=${filename}.raw.bam
O=${filename}.rmdup.bam
M=${filename}.rmdup.log
# 实时监测我们的数据发生了什么变化
samtools index ${filename}.rmdup.bam
samtools flagstat ${filename}.rmdup.bam > ./${filename}.rmdup.stat
REMOVE_DUPLICATES=true: 表示将检测出的PCR duplication直接去除;
I: 或者INPUT指定输入的bam文件;
O: 或者OUTPUT指定输出去除PCR duplication的bam文件;
M: 或者METRICS_FILE指定输出的matrix log文件。
- 下面是再次进行质控
samtools view -h -f 2 -q 30 ${filename}.rmdup.bam
| grep -v -e "mitochondria" -e "*" -e "chloroplast"
| samtools sort -O bam -@ 10 -o - > ${filename}.last.bam
# 实时监测我们的数据发生了什么变化
samtools index ${filename}.last.bam
samtools flagstat ${filename}.last.bam > ./${filename}.last.stat
-h: 输出的sam文件带header,默认不带
-f: 输出含有所有flag的reads
-q 比对的最低质量值,一般20/30都可以
其他参数介绍看 samtools 使用简述,需要说明的是由于比对到线粒体和叶绿体的reads不是我们关注的重点,因此使用grep剔除掉,当然了,做动物的话需要剔除什么视情况而定了。
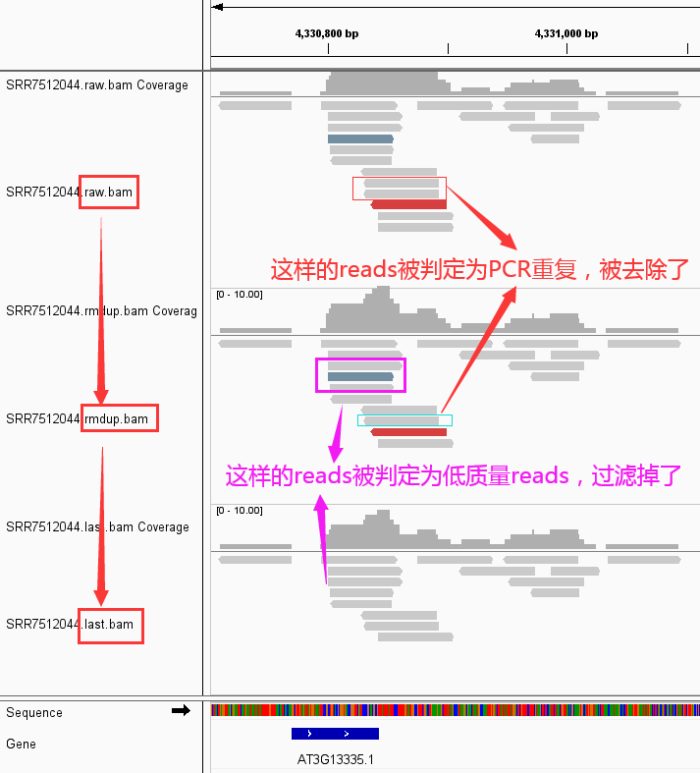
- 好了,下面来看看我们的数据从 raw data -> rmdup -> last 到底发生了那些变化
- 然后,从IGV看看发生了什么变化
关于使用IGV查看bam文件中reads呈现不同颜色的说明
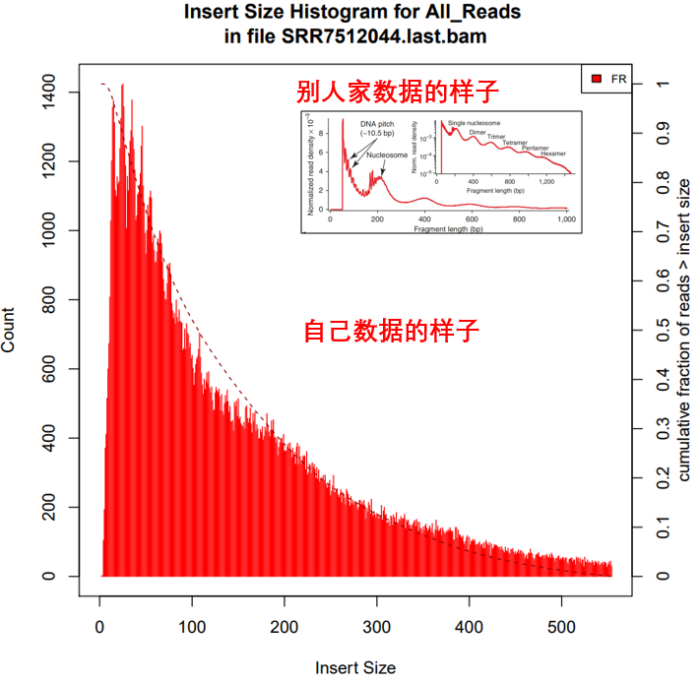
5. 统计过滤后的结果中Insertsize长度分布
- 所需软件:picard
Insertsize =(read2的比对位点 - read1的比对位点) read2的长度。也可以随便点击上图IGV中任一一条reads查看插入片段的大小
picard CollectInsertSizeMetrics
I=./${filename}.last.bam
O=./${filename}.last.insertsize
H=./${filename}.last.hist.pdf
5. peak calling与FRiP_score/SPOT_score的计算
effective_genome_size=119481543
bedtools bamtobed -i ${filename}.last.bam > ${filename}.last.bed
macs2 callpeak
-t ${filename}.last.bed
-g ${effective_genome_size}
--nomodel --shift -100 --extsize 200
-n ${filename}.last
-q 0.01 --outdir ./peaks
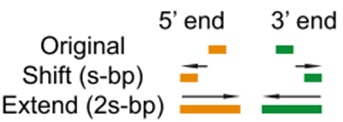
- 其实,这里面讨论最多的是
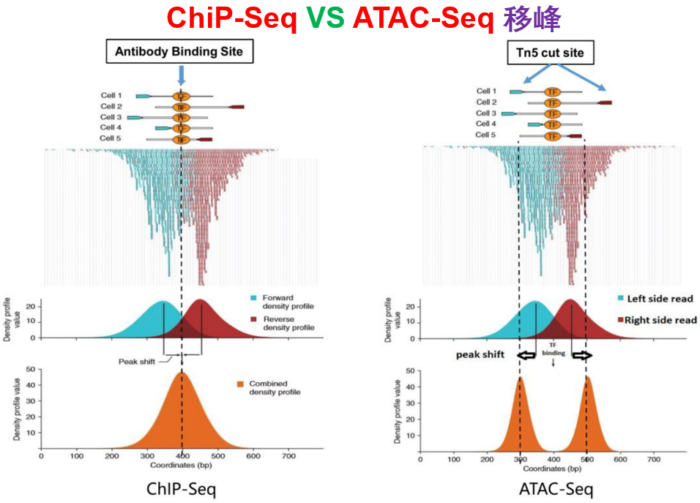
--nomodel --shift -100 --extsize 200这些参数如何选择,下面的图很形象的展示了参数的作用。当然,我也是查阅了很多资料与文献,一般默认在ATAC-seq,DNase-seq,FAIRE-seq的时候将shift设置为extsize的一半,且参数固定为:--nomodel --shift 100 --extsize 200。而在MNase-seq的时候,参数固定为:--nomodel --shift 37 --extsize 73。在ChiP-seq的时候不用移峰,所以只使用-nomodel,当做组蛋白修饰的时候,由于peak并不典型,所以使用–nomodel –shift 73参数。另外如何使用macs2 predictd预测双峰模型,这里就不展开介绍了,可以私聊我,我把脚本发你。
- 下面是我见过的图中解释移峰现象最清楚的了,可以参考
- 最后,就是输出文件的解读
- ${filename}_peaks.xls
前几行是callpeak时的命令。后面是: 1.染色体号 2.peak起始位点 3.peak结束位点 4.peak区域长度 5.peak的峰值位点(summit position) 6.peak 峰值的高度(pileup height at peak summit, -log10(pvalue) for the peak summit) 7.peak的富集倍数(相对于random Poisson distribution with local lambda)
- ${filename}_peaks.narrowPeak
narrowPeak文件是BED6 4格式,可以上传到IGV查看 1.染色体号 2.peak起始位点 3.peak结束位点 4.peak name(样本名 _peak_1的后缀之类的) 5.-10*log10qvalue 6.正负链 7.fold change 8.-log10pvalue 9.-log10qvalue 10.相对于峰的峰顶位置(relative summit position to peak start)
- 注意:excel里的起始坐标需要减1才与narrowPeak的坐标一致,见下图。
- ${filename}_summits.bed
BED格式的文件,包含peak的summits(峰顶)位置 1.染色体号 2.峰的峰顶起始位点(注意是峰顶,不是峰,他只是一个点) 3.峰的峰顶起始位点(因此二者就差一个碱基的位置) 4.峰顶(样本名 _peak_1的后缀之类的) 5.-log10pvalue
- 注意:excel里的起始坐标需要减1才与narrowPeak的坐标一致,见下图。
- 另外,需要告诉你的是,bed文件中第2列中的峰顶起始位点是如何计算的?
bed文件中第2列(峰的峰顶起始位点) = xls第二列 xls第十列 -1。或者↓↓↓↓
bed文件中第2列(峰的峰顶起始位点) = narrowPeak第二列 narrowPeak第十列 - 总览比较:
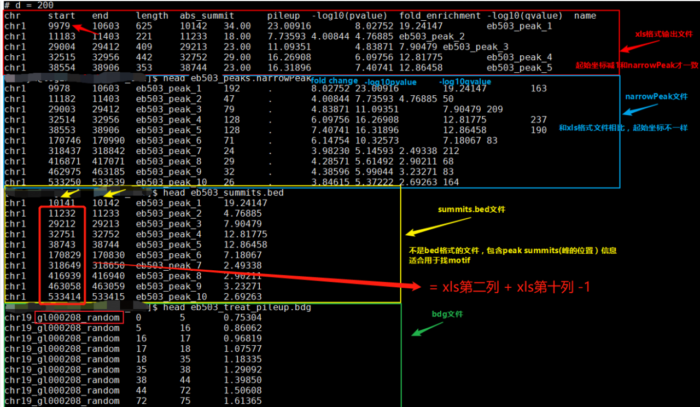 bed,narrowPeak,bdg,xls四种类型输出文件的比较
bed,narrowPeak,bdg,xls四种类型输出文件的比较 - FRiP_score的计算【官方介绍】
简单来说,FRiP_score说的就是统计落在Peaks里面的reads数量与所有比对到基因组上reads数量的比值。可以作为判定样本测序质量的一个指标。
Reads=$(bedtools intersect -a ${filename}.last.bed -b ${filename}_peaks.narrowPeak |wc -l|awk '{print $1}')
totalReads=$(wc -l ${filename}.last.bed|awk '{print $1}')
echo ${Reads} ${totalReads} ${filename} 'FRiP_score:' $(echo "scale=2;100*${Reads}/${totalReads}"|bc)'%'
- 官网上说ATAC-Seq的FRiP score should be >30%(https://www.encodeproject.org/atac-seq/)我猜是如果大多数reads都不在peaks区域,说明测序质量或者实验效果不是太好,因为酶切切开的就是peaks区域,太少不是就偏离实验目的了嘛
- SPOT_score的计算【官方介绍】【软件安装】
简单来说,SPOT_score就是在FRiP_score的基础上增加了测序深度的条件限制,比如FRiP是统计全局的reads,而SPOT只统计测序深度为30×的reads,大于30×的reads不再参与统计。目前我还在探索中如何计算。
二、高级分析
所谓的高级并不高大上,做了有可能也不能让你的文章提升档次,望周知!
1. IGV可视化
- 前面的bam文件,narrowPeak文件,bed文件都有了,现在还差一个bw文件,因此我们先跑下面的代码,然后在IGV里面统一看一下
- 所需软件:deeptools和IGV
主要是为生成bw文件用于IGV展示
bamCoverage --bam ${filename}.last.bam -o ${filename}.last.bw
--binSize 10
--normalizeUsing RPGC
--effectiveGenomeSize ${effective_genome_size}
--ignoreForNormalization chrX
--extendReads
注:如果发现没有这个包pysam.libchtslib,直接安装pip install numpydoc
-o: 生成bw文件
binSize: 自行设定覆盖度计算的窗口大小(bin)
normalizeUsing: 数据准化方法,有三种RPGC,RPKM,CPM,BPM
ignoreForNormalization: 设置你想忽略的染色体名称
extendReads: 让reads扩展至fragment的大小
- IGV一览
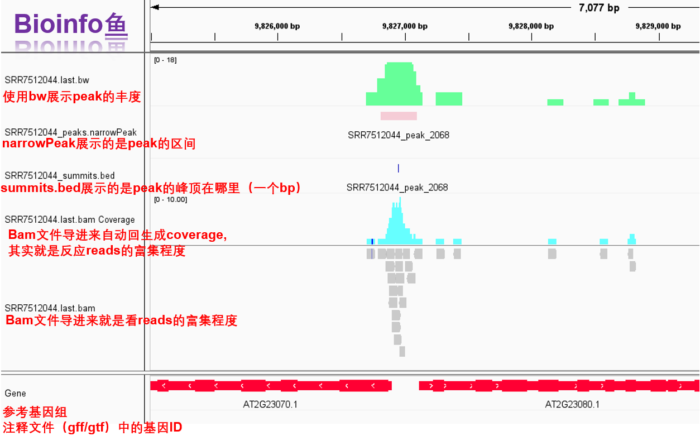
2. TSS/TES 可视化
- 所需软件:deeptools
主要是如何生成Refseq文件,虽然官网上提供了一些,但总归是不能覆盖所有物种,因此,我们可以从gff/gtf文件中自己创造。 - 先提取基因的位置信息(不是cds或者exon),根据不同的gff进行修改,但格式始终是
chrom/chromStart/chromEnd/name/score/strandawk -vFS='[\t=;]' -vOFS="\t" \ '{if ($3=="gene") print $1,$4,$5,$10,$6,$7}' \ TAIR10.gff|sed 's/Chr//g' > refseq.bed #由于tair官网上提供的基因组文件中染色人命名没有Chr,为保持一致,这里去掉
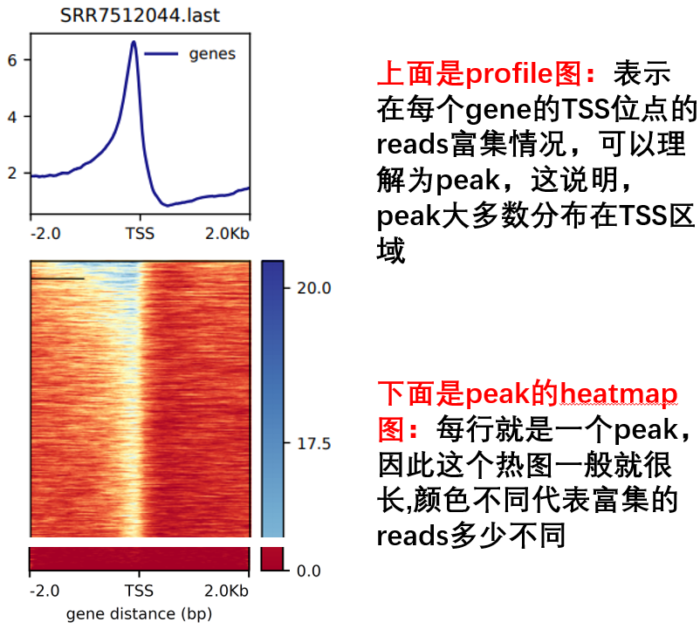
- 然后进行TSS可视化
computeMatrix reference-point --referencePoint TSS -p 15
-b 2000 -a 2000
-R ./refseq.bed
-S *.bw
-o TSS.gz
--skipZeros
--outFileSortedRegions Heatmap1sortedRegions.bed
plotHeatmap -m TSS.gz -out TSS.Heatmap.pdf --plotFileFormat pdf
plotProfile -m TSS.gz -out TSS.Profile.pdf --plotFileFormat pdf --perGroup
- 然后进行TES 可视化
computeMatrix scale-regions -p 15
-b 2000 -a 2000
-R ./refseq.bed
-S *.bw
--skipZeros -o body.gz
plotHeatmap -m body.gz -out body.Heatmap.pdf --plotFileFormat pdf
plotProfile -m body.gz -out body.Profile.pdf --plotFileFormat pdf --perGroup
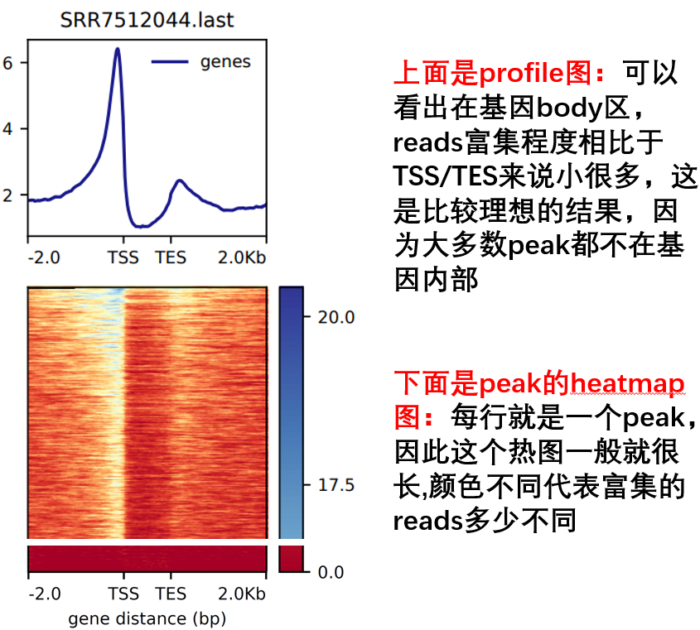
- 另外,参考下面这个图,看每个参数控制的哪里,如何同时绘制多个等。这是我好久之前总结的,清晰度堪忧
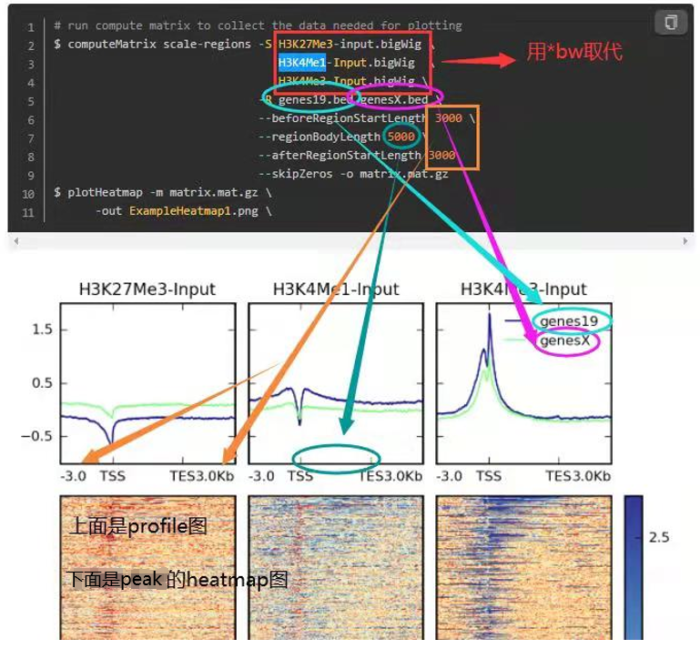
3. 相关性可视化(依旧是使用deeptools)
- 因为我测试样本少,所以就使用官网的图了哈,但代码是能跑通的,放心使用。
- 先使用 [bw] 文件生成 [npz] 文件。所有样本做相关性,用 [*.last.bw],多个单列出来也行
multiBigwigSummary bins -b *.last.bw -o number.of.bins.npz
- 画相关性热图
plotCorrelation -in number.of.bins.npz --corMethod pearson --skipZeros --plotTitle "Pearson Correlation of Average Scores Per Transcript" --plotFileFormat pdf --whatToPlot heatmap --colorMap RdYlBu --plotNumbers -o heatmap_PearsonCorr_bigwigScores.pdf --outFileCorMatrix PearsonCorr_bigwigScores.tab
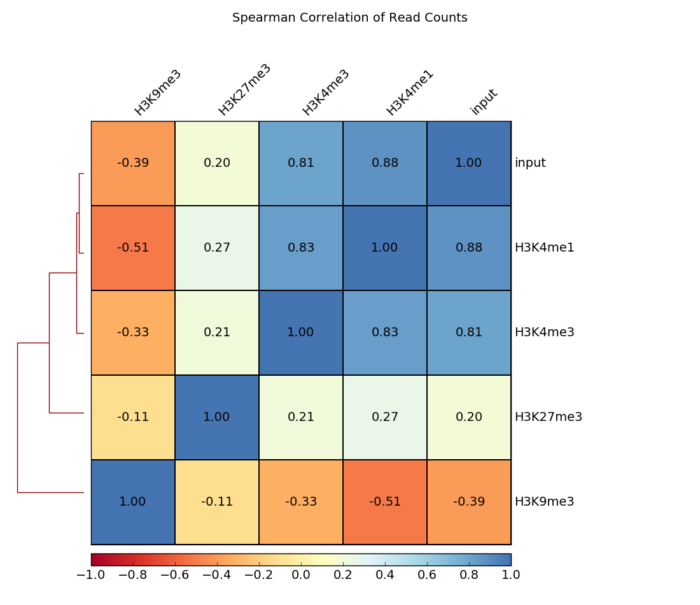
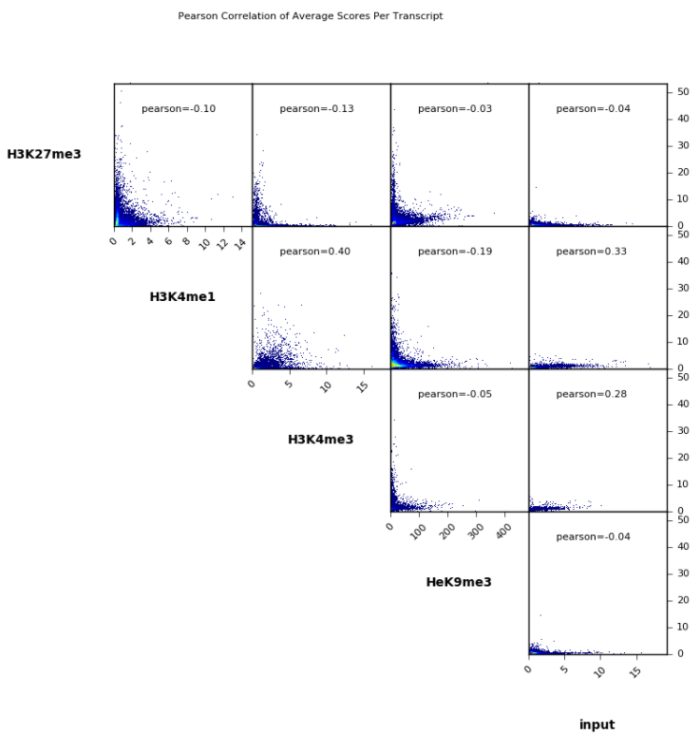
- 画相关性点图
plotCorrelation -in number.of.bins.npz --corMethod pearson --skipZeros --plotTitle "Pearson Correlation of Average Scores Per Transcript" --plotFileFormat pdf --whatToPlot scatterplot -o scatterplot_PearsonCorr_bigwigScores.pdf --outFileCorMatrix PearsonCorr_bigwigScores.tab
5. ChIPseeker对peaks进行注释和可视化
- 对peak的注释分为两个部分——结构注释和功能注释
结构注释会将peak所落在基因组上的区域结构注释出来,比如说启动子区域,UTR区域,内含子区域等等。同时,也会将与peak最临近的基因注释出来,非常好用。
功能注释其实并不是对peak进行注释的,而是对peak最临近的基因注释的,因此这部分就和传统的GO/KEGG等分析如出一辙啦。
5.1 结构注释
5.1.1 准备工作:导入包,没有的安装吧。另外需要使用gff/gtf构建一个TxDb。因为官网上TxDb也不是每一个物种都有(目前共43个),因此我们自己手动构建。
#if (!requireNamespace("BiocManager", quietly = TRUE))
# install.packages("BiocManager")
#BiocManager::install("clusterProfiler")
library(clusterProfiler)
library(ChIPseeker)
library(GenomicFeatures)
txdb <- makeTxDbFromGFF("public/bioinfoYu/genome/tair10/TAIR10.gff",
format="gff") #也可以使用gtf
keytypes(txdb) #感兴趣的话,可以用以下方法探索txdb都包含了什么内容
keys(txdb)
5.1.2 单个文件的结构注释(不推荐在这一步加OrgDB的内容,没用)
#读入单个summits文件
peaks <- readPeakFile("SRR7512044_summits.bed")
#结构注释
peakAnno <- annotatePeak(peaks,
TxDb=txdb,
tssRegion=c(-1000, 1000))
上面的tssRegion取值有说法的,因为判定"Promoter", "Exon", "Intron", "Intergenic"等结构的依据就是根据范围来的。比如定义1000以内的,则在1kbp之内规定为"Promoter (<=1kb)",而1k-2k内定义为"Downstream (1-2kb)"。但你定义2000以内呢,则在1kbp之内规定为"Promoter (<=1kb)",而1k-2k内定义为"Promoter (1-2kb)"。虽然,ChIPseeker会标记出来区域,但还是三思后再取范围。
#注释完,进行可视化,多种图可供选择
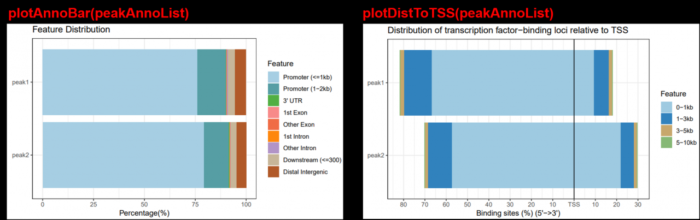
plotAnnoBar(peakAnno)
plotDistToTSS(peakAnno)
vennpie(peakAnno)
plotAnnoPie(peakAnno)
#install.packages("ggupset")
library(ggupset)
upsetplot(peakAnno)
#install.packages("ggimage")
library(ggimage)
upsetplot(peakAnno, vennpie=TRUE)
#最后将我们的注释结果转为数据框,便于查看
df <- as.data.frame(peakAnno)
#将注释到的基因提取出来(第14列),用于后续功能分析
gene <- df[,14]
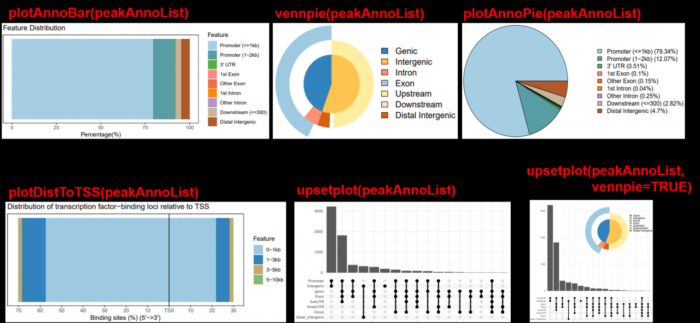
5.1.3 多个文件的结构注释
#一次也可以读入多个summits文件,使用list存储,然后使用lapply注释
files = list(peak1 = "peaks.bed", peak2 = ("peak.bed"))
peakAnnoList <- lapply(files,
annotatePeak,
TxDb=txdb,
tssRegion=c(-2000, 2000))
plotAnnoBar(peakAnnoList)
plotDistToTSS(peakAnnoList)
5.2 功能注释
5.2.1 可选支线
由于这里需要物种的OrgDb注释库。如果你足够幸运,研究的物种正好在官网仅提供的20个物种中存在,那就拿来用吧。除了以上这些都好说,还有是id转换,后面再说。
5.2.2 OrgDb下载
本次测试是用拟南芥的样本,我推荐在官网中直接将所需要的的OrgDb库下载到服务器上,这样就不用在R里面重复在网上获取浪费时间了。比如说拟南芥,就可以下载,这样就把【org.At.tair.db】下载到了服务器或者本地了。
wget http://www.bioconductor.org/packages/release/data/annotation/src/contrib/org.At.tair.db_3.13.0.tar.gz tar -zvxf org.At.tair.db_3.13.0.tar.gz
5.2.3 导入OrgDb
#从本地下载OrgDb库(如果你直接下载到了R的lib里面,就直接library也可)
install.packages("/public/bioinfoYu/genome/tair10/org.At.tair.db", repos=NULL)
#另外一种方法
#if (!requireNamespace("BiocManager", quietly = TRUE))install.packages("BiocManager")
#BiocManager::install("org.At.tair.db")
#载入库
library(org.At.tair.db)
#感兴趣的话,看看org.At.tair.db包含的都是些啥
#查看包含的列名
keytypes(org.At.tair.db)
#查看包含的基因ID
keys(org.At.tair.db)
#随便选择一个基因和一些列名,看一看到第是什么东东,不过因为导入了其他包,下面命令报错,我至今还没有解决
select(org.At.tair.db,keys="AT1G01010",columns = c("ENTREZID","GO","SYMBOL"))
5.2.4 ID转换(非必须)
上面5.1.2已经得到peak附近的基因集【gene】了,如果需要ID转换,就是用clusterProfiler中的bitr函数
#首先,去除版本号,如AT1G50040.1后面的.1,当然我们的测试集没有,因此不用做
#gene_rm <- gsub(".[0-9] $","",gene)
#下面进行ID转换
keytypes(org.At.tair.db)
gene_last <- bitr(geneID = gene,
fromType = "TAIR",
toType = c("ENTREZID", "SYMBOL", "GENENAME"),
OrgDb = org.At.tair.db)
# geneID 需要转换的ID
# fromType 当前ID类型
# toType 转换成什么ID,使用keytypes()查看有哪些类型
# OrgDb 注释数据库
5.2.5 GO功能注释
# 因为keyType不接受数据框类型的数据,因此我们转换为字符型
genelist <- gene.symbol[,2] #注意我这里把ENTREZID单独拿出来了,所以下面自然是keyType = "ENTREZID",
go_result <- enrichGO(genelist,
keyType = "ENTREZID",
OrgDb = org.At.tair.db,
ont = "BP", #BP,MF,CC,ALL可选
pAdjustMethod = "BH",
qvalueCutoff = 0.05,
readable = FALSE)
#可视化,showCategory=20调整显示的条目多少
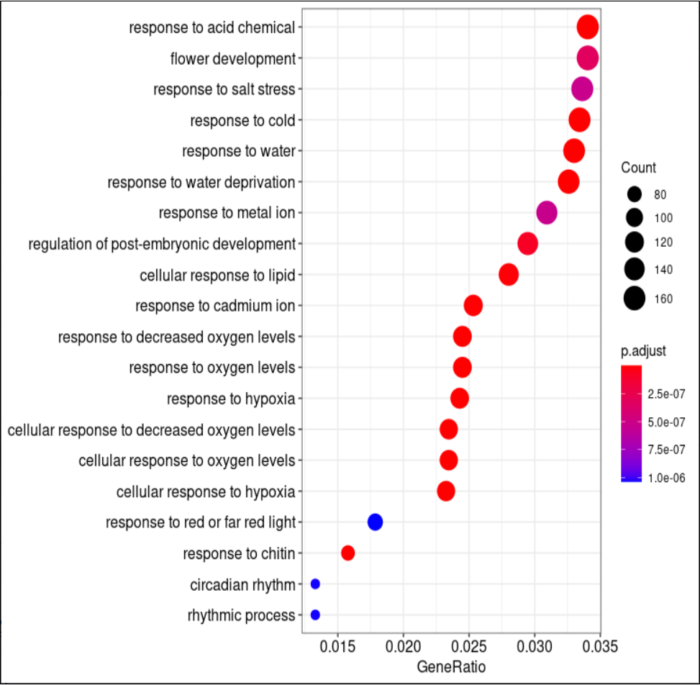
dotplot(go_result, showCategory=20)
#将结果转为数据框,可以选择导出
goresult <- as.data.frame(go_result)
5.2.6 KEGG通路注释
#geneClusters必须是数据框类型,ID必须是ENTREZID类型
class(gene.symbol)
compKEGG <- compareCluster(geneClusters=gene.symbol,
fun = "enrichKEGG",
organism = "ath", #在 http://www.genome.jp/kegg/catalog/org_list.html 上查看物种简写
pvalueCutoff = 0.05,
pAdjustMethod = "BH")
dotplot(compKEGG, showCategory = 20, title = "KEGG Pathway Enrichment Analysis")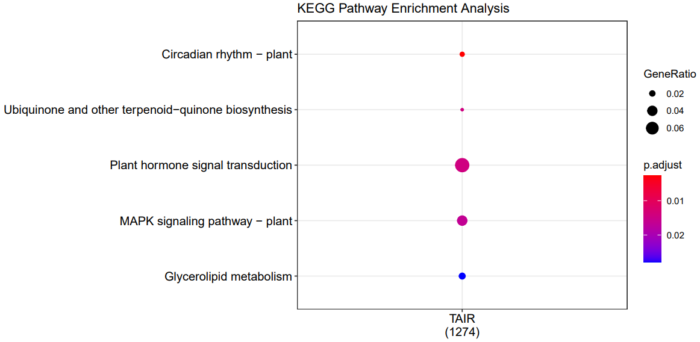
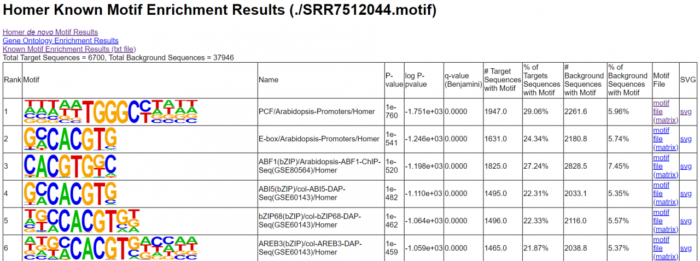
6. homer寻找motif
# 提取peaks的位置信息文件
awk '{print $4"t"$1"t"$2"t"$3"t "}'
SRR7512044_peaks.narrowPeak > SRR7512044.homer_peaks.tmp
# 寻找motif
findMotifsGenome.pl SRR7512044.homer_peaks.tmp
./TAIR10_genome.fa #指定参考基因组
./SRR7512044.motif #输出文件夹的名字
-size 200 -len 8,10,12
7. DiffBind找差异peak
- 所需软件:DiffBind
首先根据我自己的理解将找差异peak的原理说一下:既然是找差异的peak,那首先就要保证不同样本间peak所在的区间基本一致才可以。因此第一步就是找到多个样本相同的peak区间。第二步,基于这些相同区域的peak开始找差异,这个地方的差异和传统的RNA-Seq找差异基因的原理一模一样,都是对落在峰区间的reads进行计数,数量不同的话差异就由此而来。但软件都帮我们做了,就不用考虑那么多了
- 我废话真多,其实最麻烦的就是准备输入数据,下面来看怎么做
一般情况相下需要包括以下几列:"SampleID", "Tissue", "Factor", "Condition" ,"Treatment","Replicate", "bamReads" ,"ControlID" ,"bamControl" ,"Peaks"和"PeakCaller"
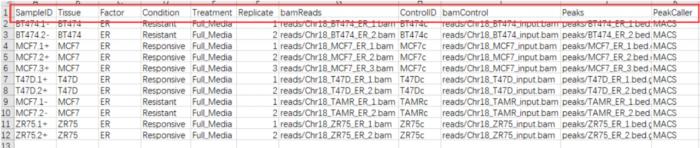
- 但我们做ATAC-Seq的哪里有什么
control,controlID,bamControl,因此删掉,而"Tissue", "Factor", "Condition" ,"Treatment","Replicate",表示的都是分组的意思,因此我保留了两个"Factor","Replicate",最后csv文件的样子如下: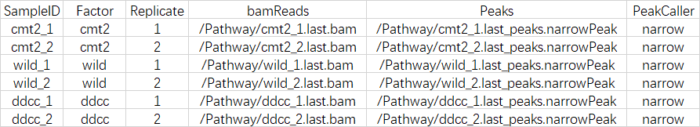
SampleID:不能有重复
Factor:进行分组,后面要做差异分析的依据
Replicate:重复的编号
bamReads:最后生成的bam文件。全路径 bam文件名
Peaks:macs2生成的narrowPeak文件。全路径 narrowPeak文件名
PeakCaller:说明peak类型,使用narrowPeak文件则为narrow。使用xls文件,则用macs。使用bed文件,则用bed
- 开始正题
# 安装包
if (!requireNamespace("BiocManager", quietly = TRUE))install.packages("BiocManager")
BiocManager::install("DiffBind")
library(DiffBind)
#导入数据,dba函数会将会将bam文件一同导入,因此csv中的路径一定要准确
tamoxifen <- dba(sampleSheet="atDiff.csv")
#查看样本间的相关性
plot(tamoxifen)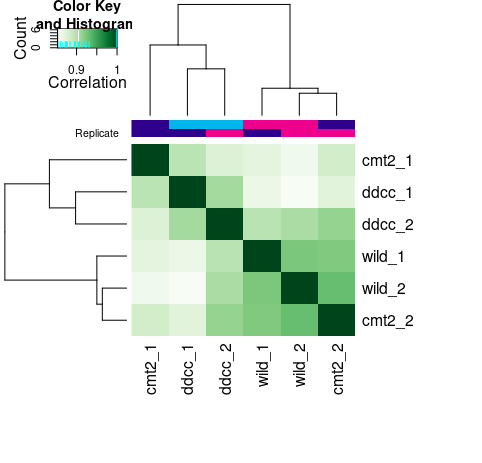
#计算每个样本中的reads数 tamoxifen <- dba.count(tamoxifen) #简单查看计算结果 info <- dba.show(tamoxifen) libsizes <- cbind(LibReads=info$Reads, FRiP=info$FRiP, PeakReads=round(info$Reads * info$FRiP)) rownames(libsizes) <- info$ID libsizes
#统计结果
LibReads FRiP PeakReads
cmt2_1 6720462 0.29 1948934
cmt2_2 7747491 0.33 2556672
wild_1 7078309 0.36 2548191
wild_2 8030752 0.34 2730456
ddcc_1 6370232 0.30 1911069
ddcc_2 6956429 0.35 2434750
<pre">#默认基于测序深度对数据进行标椎化 tamoxifen <- dba.normalize(tamoxifen) norm <- dba.normalize(tamoxifen, bRetrieve=TRUE) norm #查看进行标准化的文库大小(library-size),其实就是各样本文库大小总和的平均值,你可以计算验证一下 normlibs <- cbind(FullLibSize=norm$lib.sizes, NormFacs=norm$norm.factors, NormLibSize=round(norm$lib.sizes/norm$norm.factors)) rownames(normlibs) <- info$ID normlibs
#标椎化结果
FullLibSize NormFacs NormLibSize
cmt2_1 6720462 0.9398443 7150612
cmt2_2 7747491 1.0834724 7150612
wild_1 7078309 0.9898885 7150612
wild_2 8030752 1.1230859 7150612
ddcc_1 6370232 0.8908652 7150612
ddcc_2 6956429 0.9728438 7150612
#分组,格式是表头在最前面,要分的组依次写在后面,只能两两比较,因此后面只能写两组,但可以多执行几次,都会追加到tamoxifen 中
#分组1,后面使用contrast=1单独查看
tamoxifen <- dba.contrast(tamoxifen, contrast=c("Factor","ddcc","wild"))
#分组2,后面使用contrast=2单独查看
tamoxifen <- dba.contrast(tamoxifen, contrast=c("Factor","cmt2","wild"))
#当然还可有分组3,4,5等,均可以使用contrast=number单独查看
tamoxifen
#按照分组分别进行差异分析,默认使用DESeq2进行计算,可以选择method = DBA_EDGER(edgR),或者两个都要method = DBA_ALL_METHODS
tamoxifen <- dba.analyze(tamoxifen)
dba.show(tamoxifen, bContrasts=TRUE)
#查看差异分析的结果与导出为csv文件
#查看第1组差异分析的结果
tamoxifen.DB_1 <- dba.report(tamoxifen,contrast=1)
tamoxifen.DB_1
write.csv(tamoxifen.DB_1, file="diffBind_result.csv")
#查看第2组差异分析的结果
tamoxifen.DB_2 <- dba.report(tamoxifen,contrast=2) tamoxifen.DB_2 write.csv(tamoxifen.DB_2, file="diffBind_result.csv") ##传统的上下调在找差异peak中称为“Gain”或“Loss” #查看第1组的差异peak数量“Fold>0或<0”控制是“Gain”或“Loss” sum(tamoxifen.DB$Fold>0,contrast=1)
sum(tamoxifen.DB$Fold<0,contrast=1) #查看第2组的差异peak数量“Fold>0或<0”控制是“Gain”或“Loss” sum(tamoxifen.DB$Fold>0,contrast=2)
sum(tamoxifen.DB$Fold<0,contrast=2)
- 由于我是使用两组进行测试的,所以可以使用contrast=1或contrast=2分别查看,由于都是相同的代码,下面进行可视化的时候,只选择contrast=1进行可视化
#韦恩图可视化
dba.plotVenn(tamoxifen, contrast=1, bDB=TRUE, bGain=TRUE, bLoss=TRUE, bAll=FALSE)
#PCA
dba.plotPCA(tamoxifen) #这里可以使用所有样本进行PCA
dba.plotPCA(tamoxifen, contrast=1, label=DBA_FACTOR)#单独对分组1进行PCA
#MA plots
dba.plotMA(tamoxifen, contrast=1)
#Volcano plots
dba.plotVolcano(tamoxifen, contrast=1)
#Box plots
dba.plotBox(tamoxifen, contrast=1)
#Heatmap
hmap <- colorRampPalette(c("red", "black", "green"))(n = 13)
#对所有样本的所有的差异peak画热图
dba.plotHeatmap(tamoxifen,correlations=FALSE,scale="row",colScheme = hmap)
#单独对分组1中所有的差异peak画热图
dba.plotHeatmap(tamoxifen, contrast=1, correlations=FALSE, scale="row", colScheme = hmap)
dba.plotHeatmap(tamoxifen,correlations=FALSE,scale="row",colScheme = hmap)
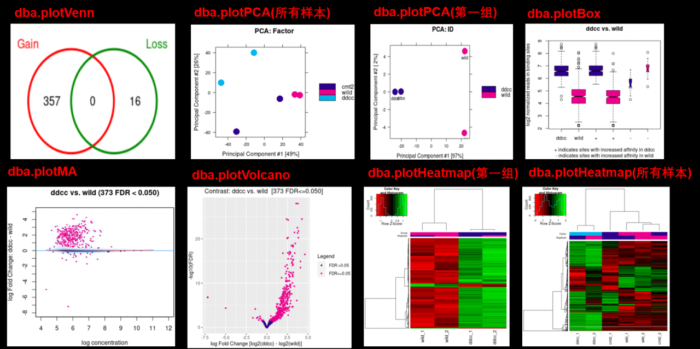
- 想画下面的图,可是一直提示could not find function dba.plotProfile,因此还没有成功,用官方提供的图展示一下吧,大家能画出来可以和我说一声哦。这是【官方文档】可以参考
#Profiling and Profile Heatmaps dba.plotProfile(tamoxifen)
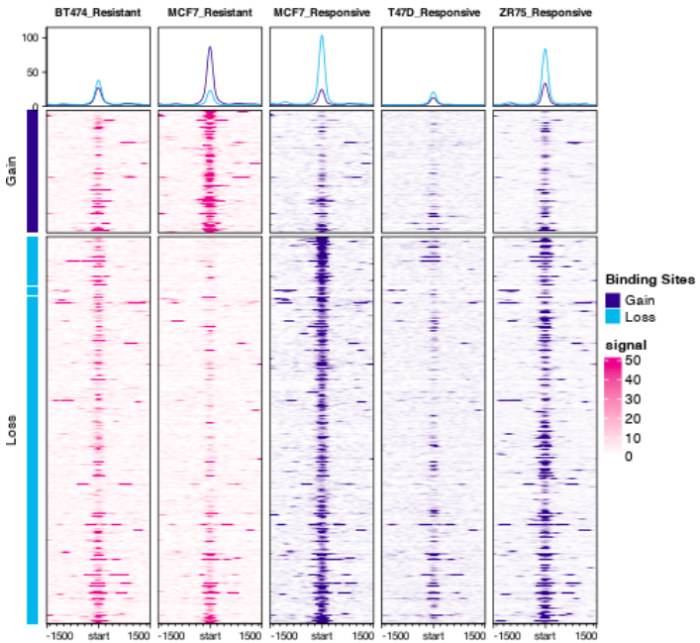
8. footprint分析
目前我还没有找到使用于所有物种的软件,HINT-ATAC用来做footprint分析的话,应该只支持不几个物种,比较鸡肋。ATACseqQC应该是一款不错的软件,最近还在探索
三、ATAC-seq与多组学数据联合分析
转录因子ChIP-seq:由于大部分转录因子结合的是染色质开放区域,所以ATAC-seq的Peak可能和转录因子ChIP-seq的Peak存在部分重叠的情况,而且ATAC-seq得到的Peak长度往往更长,因此ATAC-seq数据和转录因子ChIP-seq数据可以相互验证。转录因子在ChIP-seq中独有的Peak暗示这个转录因子可能是结合在异染色质区域的驱动型转录因子(Pioneer TFs),驱动型转录因子随后招募染色质重塑复合体以及其它转录因子开始转录。另外,联合分析已经报道的ChIP-seq数据可以更准确地分析转录因子的足迹。
组蛋白修饰ChIP-seq:ATAC-seq数据同样可以和组蛋白修饰ChIP-seq数据进行联合分析,其中转录激活性修饰(H3K4me3,H3K4me1和H3K27ac等)与染色质开放程度呈正相关,转录抑制性修饰(H3K27me3)与染色质开放程度呈负相关。联合已知的增强子和启动子之间的相互作用数据也可以帮助构建调控网络。
RNA-seq:ATAC-seq数据可以通过联合分析RNA-seq数据来发现哪些差异表达的基因是受染色质可及性调控的,进一步可以推测这些差异表达的基因哪些是受开放染色质中具有motif和footprint的转录因子调控的,因此ATAC-seq与RNA-seq的联合分析有助于破译基因调控网络和细胞异质性。