

之前分析了好几个公共项目,拿到的peaks都很诡异,搞得我一直怀疑是不是自己分析错了。终于,功夫不负有心人,我分析了一个数据,它的peaks非常完美!!!可以证明,我的分析流程以及peaks绘图代码并没有错!数据来自于http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE74311,是关于H3K27ac_ChIP-Seq_LOUCY,组蛋白修饰的CHIP-seq数据,很容易就下载了作者上传的测序数据,然后跑了我的流程!https://github.com/jmzeng1314/NGS-pipeline/tree/master/CHIPseq
本文的重点在于讲解如何查看自己的peaks是否是正确的!我是直接用比对的bam文件来用samtools depth命令来获取peaks区域的测序深度,从而画图的,代码见step5-peaks-view-samtools-depth.R
在终端调用我的代码画图命令如下;
Rscript ~/scripts/peakView.R ../unique_peaks.bed ../../SRR2774675.unique.sorted.bam ../../SRR2774676.unique.sorted.bam Rscript ~/scripts/peakView.R ../unique_peaks.bed ../../SRR2774675.unique.sorted.bam ../../SRR2774676.unique.sorted.bam
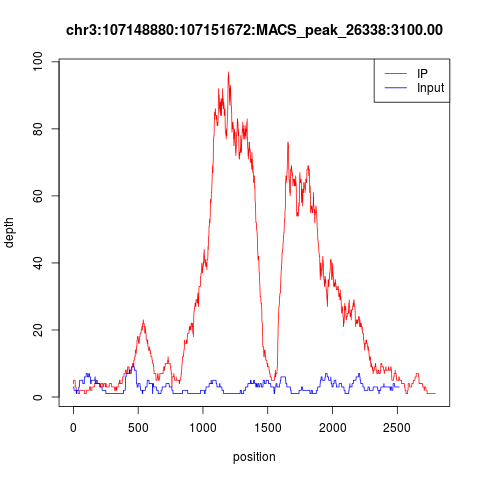
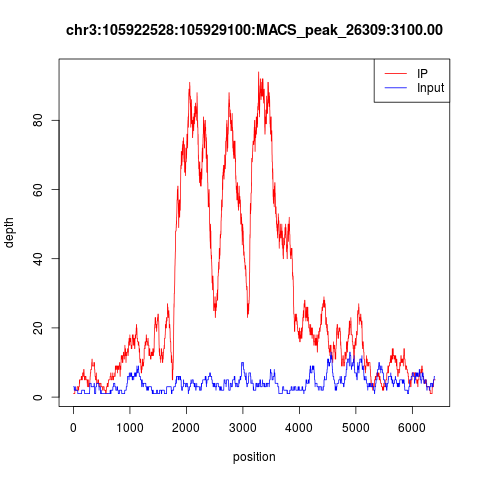
下面随便看两个peaks,很明显是双峰模型,而且IP的测序深度远高于INPUT,数据非常棒!
然后我不得不指出如果CHIP-seq实验失败,那么peaks会很诡异,首先你会看到测序深度大多都在10以下,即使有部分测序深度很高的,也是IP和INPUT的测序深度压根就没有差异,下面就是一个典型的失败案例!
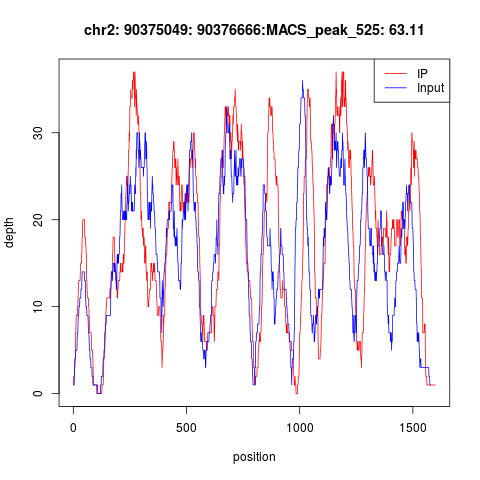
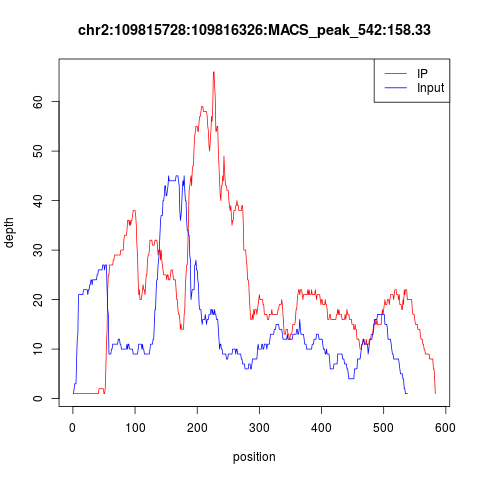
这种实验失败的数据,实在是无法分析。而转录因子的CHIP-seq实验失败率还挺高的,所以一定要有control
原文来自:http://www.bio-info-trainee.com/1843.html
1F
我是原作者,发现你转载了我不少文章,请联系一下我,邮箱你知道的。