

bsseq 主要用来分析WGBS的数据, 安装过程如下
source(“http://bioconductor.org/biocLite.R“) biocLite(“bsseq”)
bsseq的分析主要包括以下4步:
- 读取原始数据
- BSmooth
- t-test检验
- DMR
1. 读取原始数据
bsseq要求的原始数据格式如下:
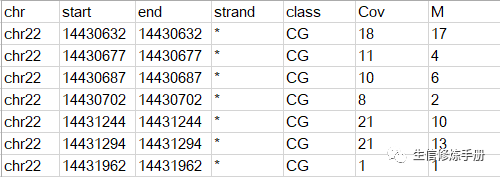
共6列数据,制表符分隔,每一行代表一个甲基化位点,前5列很好理解,描述甲基化位点的染色体位置和类别,默认情况下bbseq用于分析CpG类型的甲基化位点。当然其他类型的数据,比如CHG, CHH也支持,但是需要调整参数。Cov代表覆盖到这个位点的reads数,M代表其中发生了甲基化的reads数目。
每个样本一个这样的原始数据,用来表示该样本methylation calling的结果,这样的数据我们从bismark的结果中也可以得到。当原始数据准备好之后,首选需要读取所有样本的原始数据,然后导入到R中,生成一个bbseq定义的对象。在bbseq安装的路径下,提供了一个名为get_BS.chr22.R的脚本,展示了如何从读取所有样本原始数据的过程。
代码如下
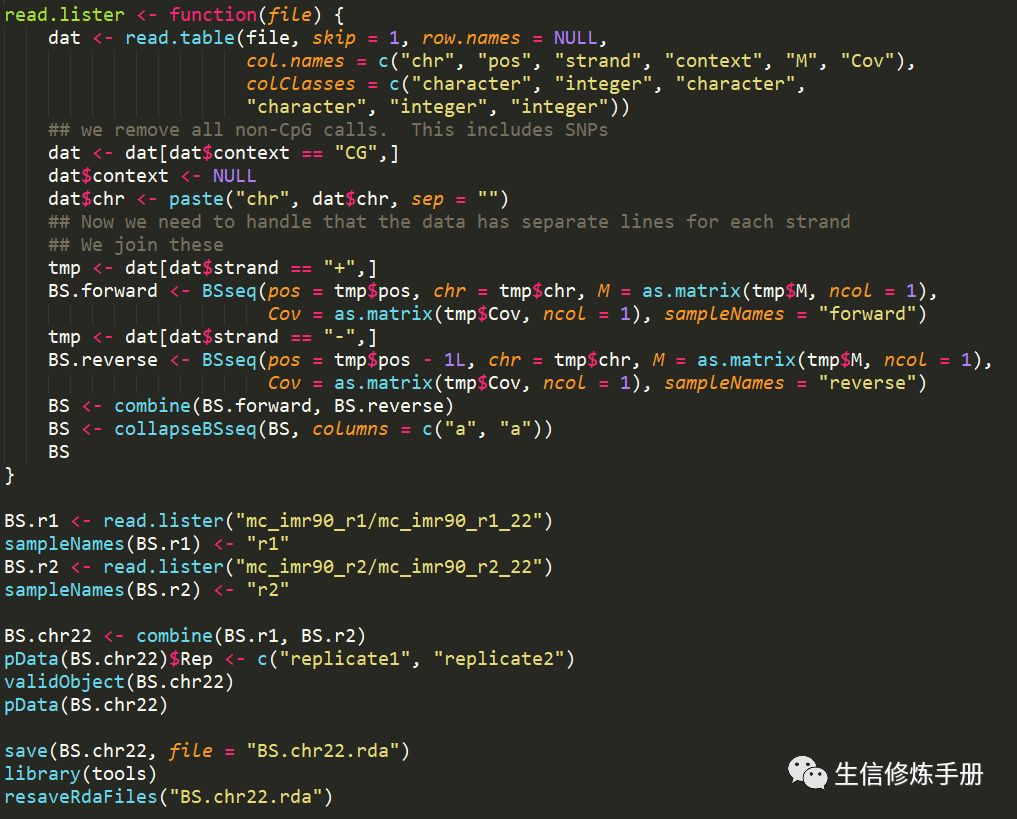
这里以mc_imr90_r1_22和mc_imr90_r2_22两个样本的原始数据为例,详细展示了读取过程。我们只需要根据自己的数据,适当修改上述代码就可以了。主要注意sampleNames和pData数据就可以了。
2 Smooth
已测试数据BS.chr22为例,smooth的过程如下
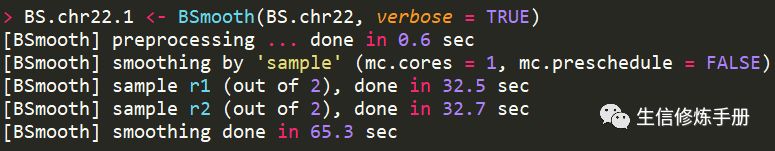
在实际分析中,由于甲基化位点很多,所以这一步时间特别久,为了提高速度,可以添加mc.cores参数,这个参数指定了CPU个数,用于并行执行。
BS.chr22.1 <- BSmooth(BS.chr22, mc.cores = 2, verbose = TRUE)
3. T-test
在分析之前,有必要过滤掉覆盖度较低的甲基化位点。通常保留在所有样本中覆盖度大于2的甲基化位点,但是也可以修改这个条件。过滤之后,直接通过BSmooth.tstat进行分析
下面的代码基于bsseqData包中的数据,这个数据包含了6个样本,分为normal和cancer两组
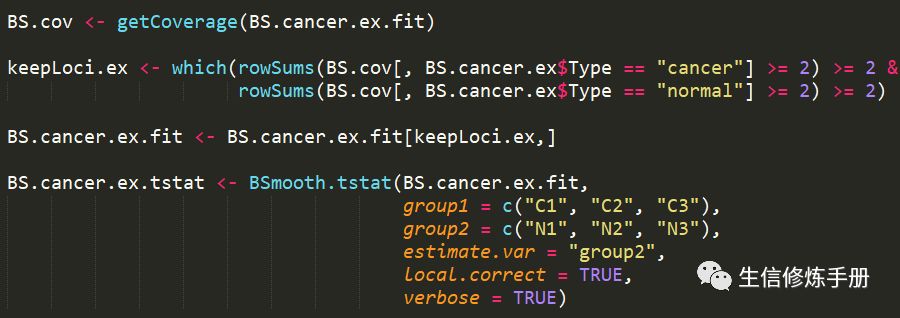
group1 指定属于treatment组的样本,group2指定属于control组的样本。
4. DMR
通过dmrFinder 函数进行差异甲基化分析, 代码如下:

cutoff 指定DMR的阈值,这个阈值根据t-test的结果进行调整。subset对差异甲基化的结果进行筛选,筛选包含甲基化位点个数大于3而且meanDiff 大于0.1的甲基化区域。
1F
methpipe处理后可以进行bsseq 差异分析吗?