

我们统一选择p<0.05而且abs(logFC)大于1的基因为显著差异表达基因集,对这个基因集用R包做KEGG/GO超几何分布检验分析。
然后把表达矩阵和分组信息分别作出cls和gct文件,导入到GSEA软件分析。
基本任务是完成这个分析
其实这一步特别的简单,就是筛选,然后用专门的R包分析就好了。但是富集分析貌似简单,但其实充满了变数。
PS: 下面的内容我直接从我之前的文章里摘录过来了。
为何要基因富集分析
在基因差异表达分析之后,你得到了好多p值特别小(也就是显著性很高)的基因,那么下一步你想做什么?
- 选择一些基因用于验证?
- 对其中基因进行后续研究?
- 在结果中把这些基因都放在后面?
- 尝试着把所有基因相关的文献都都读读看(劝你放弃这个念头)?
- 欢迎补充
这些想法都是非常顺理成章的,但是不要着急。
首先,差异表达找到的基因往往很多,你简单的粗暴去找每一个基因的详细资料,显然不太现实;
其次,如果我们单纯觉得某一个基因和你研究的课题相关,或者说你其实已经找到了一个有可能的基因(或者你只是希望用一些高大上的实验验证一下)那么这个行为是不是有太多主观性,存在一些偏见。
当然,你觉得基因就是你要找的,可是万一它只是碰巧来打酱油的呢,这不就是很尴尬了。
所以为了让审稿人相信你的结果,你就需要做一个基因富集分析哦。
什么是基因富集分析
基因富集分析(gene set enrichment analysis)是在一组基因或蛋白中找到一类过表达的基因或蛋白。一般是高通量实验,如基因芯片,RNA-Seq,蛋白质组学(质谱结果)的后续步骤。
基因富集分析需要我们提供某一类功能基因的集合用于背景,常用的注释数据库如:
- The Gene Ontology Consortium: 描述基因的层级关系
- Kyoto Encyclopedia of Genes and Genomes: 提供了pathway的数据库。
分析方法
在文献Ten Years of Pathway Analysis: Current Approaches and Outstanding Challenges(推荐大家看一遍)作者将研究方法归为三种,其中第三种方法想的很好就是难度很大。:
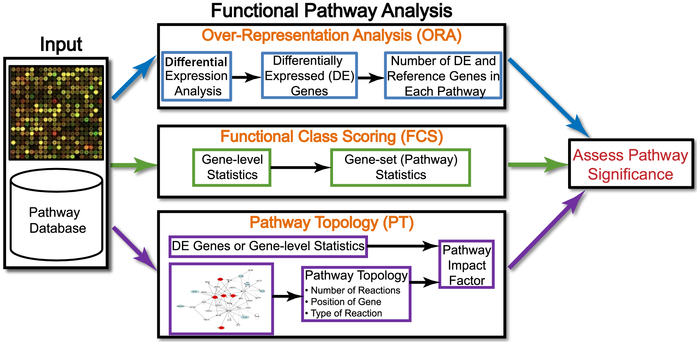
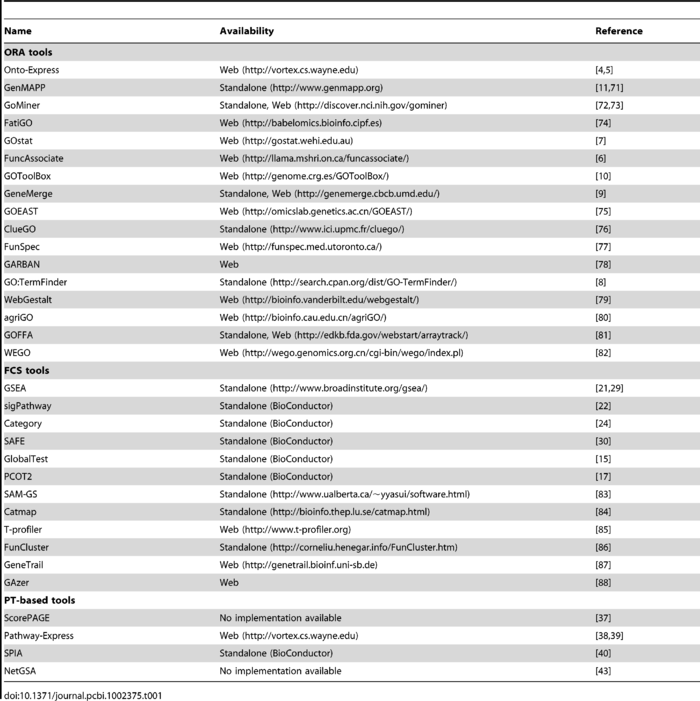
作者还贴心的把每一种方法有哪些工具都总结出来了:
Over-Repressentation Analysis(ORA)
ORA是目前商业化最多的方法。为了说明他的基本思想,我要举一个喜闻乐见的例子:读书无用论。
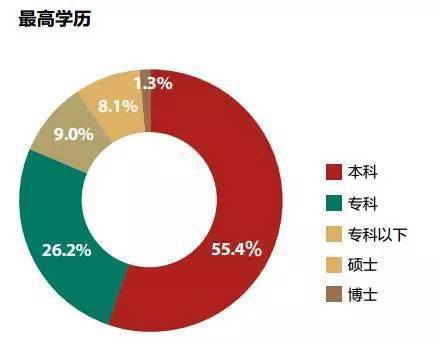
这是我百度找到搜狐财经一篇文章《大数据告诉你真正的有钱人是什么样》的有钱人的学历分布情况,高学历人群(本科以上,因为本科生太多了)所占的比例是9.4%,其他都是一般学历占90.6%。这时候,有些公众号就可以开始不带脑子的说了,读书没什么用呀,有钱人中都是一般学历的呀,以后读书读到大学就行了,甚至也可以不上本科呀(34.2%本科和本科以下)。
你每年回家总能回去看到有人炫耀说,虽然我有钱,可是读书太少了,都不能和你们读书人比的。你总感觉哪里不对劲,但是却又不太方便说出来。
实际上,这就是因为没有考虑到背景。因为高学历本身人数就不多,当然在有钱人里面的人数也就相应不多了。我们要证明有钱人更多是富集高学历这一部分。
| 类别 | 有钱人 | 普通人 |
|---|---|---|
| 高学历 | 10 | 50 |
| 一般学历 | 90 | 850 |
| 100 | 900 |
H0: 是否有钱和学历高无关
Ha: 学历高还是有点用的
然后做一个Fisher精确检验,看看p值。
richer.pop <- matrix(data = c(10,90,50,850),nrow=2)
fisher.test(richer.pop, alternative = "greater")
Fisher's Exact Test for Count Data
data: richer.pop
p-value = 0.03857
alternative hypothesis: true odds ratio is greater than 1
95 percent confidence interval:
1.052584 Inf
sample estimates:
odds ratio
2.109244p值小于0.05,看来我读个博士让我以后有钱概率变大了。
现在将我们上面的有钱人改成我们找到的基因,整体改成所有基因。高学历表示属于目标注释基因集,一般学历就是非注释基因组.我们就是要判断我们找到的基因更多是在目标注释集中。所以你需要列出下表,然后再做一个fisher.test()。
| 类别 | 感兴趣的基因 | 其他基因 |
|---|---|---|
| in anno group | 10 | 50 |
| not in anno group | 290 | 19950 |
| 300 | 20000 |
上述的基本思想就是统计学的白球黑球实验:
在一个黑箱里,有确定数量的黑白两种球,你随机抽取(不放回)M个球中,其中两种球的比例分别是多少?
除了用Fisher精确检验,还有其他统计方法:
- Hypergeometric (fisher精确检验用的就是超几何检验)http://www.bio-info-trainee.com/1225.html
- Binomial: 二项分布要求是有放回,无放回要求整体足够大大到可以近似。
- Chi-squared
chisq.test(counts) - Z
- Kolmogorov-Smirnov
- Permutation http://www.bio-info-trainee.com/1237.html
ORA的方法就是如此的简单,但是有一个问题,就是你如何确定哪些基因是差异表达的,你还是需要设置一个人为的cutoff, 主观能动性成分有点大。
Functional Class Scoring(FCS)
FCS认为,“虽然个体基因表达改变之后会更多在通路中体现,但是一些功能相关基因中较弱但协调的变化也有明显的影响。”
The hypothesis of functional class scoring (FCS) is that although large changes in individual genes can have significant effects on pathways, weaker but coordinated changes in sets of functionally related genes (i.e., pathways) can also have significant effects
FCS分析方法稍微复杂了一点,他要求的输入是一个排序的基因列表和一个基因集合。MIT, Broad Institute 2007年文献就提供了这一方法的软件"GSEA"
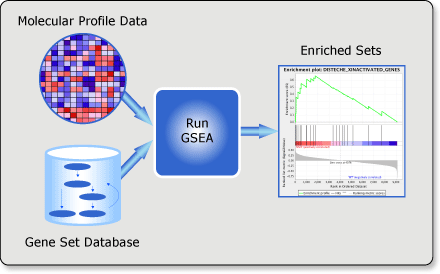
有如下特点:
- 计算所有输入基因集合的分数,而不是单个基因
- 不需要设置cutoff
- 找到一组相关的基因
- 提供了更加稳健的统计框架
GSEA是一款图形化的软件,根据他们提供的教程,然后点呀点,就会得到如下结果。下图就是需要好好理解的部分。
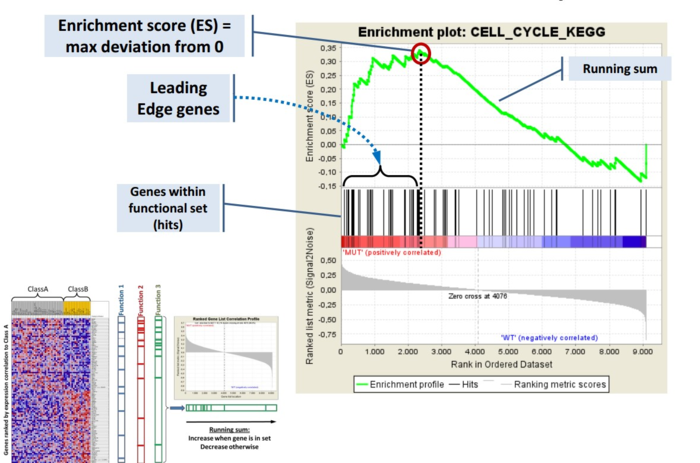
中间从蓝色到红色的过渡“带”表示基因从上调到下调排列(排序可以按照fold change,也可以是p-value)。黑色像条形码的竖线表示该位置的基因属于某个指定通路。绿色有波动的曲线表示富集分数,从0开始计算,属于基因通路增加,不属于则减少。最后看下黑色的条形码是不是富集在一端。
那如何做统计检验呢?
The final step in FCS is assessing the statistical significance of the pathway-level statistic. When computing statistical significance, the null hypothesis tested by current pathway analysis approaches can be broadly divided into two categories: i) competitive null hypothesis and ii) self-contained null hypothesis [3], [18], [22], [31]. A self-contained null hypothesis permutes class labels (i.e., phenotypes) for each sample and compares the set of genes in a given pathway with itself, while ignoring the genes that are not in the pathway. On the other hand, a competitive null hypothesis permutes gene labels for each pathway, and compares the set of genes in the pathway with a set of genes that are not in the pathway。
我们要检验的目标是基因富集在一端是因为于目标通路相关的基因都在一端富集。那么空假设就是,你把找到的基因随便摆放也能看到富集现象。用比较专业的话说就是先生成一个零假设的数据分布,然后观察实际数据在这个零假设分布下,是不是在尾端 。
一些问题
统计检验的能力是有限的,所以还有很多问题存在解决。
- 我们希望找到生物学显著的基因,但是生物学显著和统计显著两者并不是完全相关
- 无论是ORA还是FCS都对背景(也就是这个物种一共有多少基因)有要求,但是随着我们的研究深入,基因数量会改变。有些软件会直接设置一个很大的背景数,从而让p值很显著,然后我们就开心地用他们的结果。
- 有些基因没有注释,也就是注释缺失,处理方法就是扔(欢迎拍砖)。
- 有一些注释项是其他项的子集。
富集分析
我们这里用来做富集分析的工具是Y叔的clusterProfiler。基本上用这个软件就对了,简单还用,而且结果正确。它支持上面提到的三类方法,所以也没有必要专门导出数据用GSEA软件了:
- Over-Representation Analysis
- Gene Set Enrichment Analysis
- Biological theme comparison
前提准备:
- 数据筛选,根据padj < 0.05 且Log2FoldChange的绝对值大于1的标准。
deseq2.sig <- subset(res, padj < 0.05 & abs(log2FoldChange) > 1)
- 安装包下载注释数据(rog.HS.eg.db)
source("https://bioconductor.org/biocLite.R")
biocLite("clusterProfiler")
biocLite("AnnotationHub")
library(AnnotationHub)
ah <- AnnotationHub()
org.hs <- ah[['AH53766']]
注释数据库根据分析的物种决定,如何查找数据库,可以看我写的一篇文章用Bioconductor对基因组注释# 用Bioconductor对基因组注释
GO富集和GESA
GO分析参考Y叔写的GO analysis using clusterProfiler文档:
主要的函数是
enrichGO(gene, OrgDb, keytype = "ENTREZID", ont = "MF", pvalueCutoff = 0.05, pAdjustMethod = "BH", universe, qvalueCutoff = 0.2, minGSSize = 10, maxGSSize = 500, readable = FALSE, pool = FALSE)
主要关注如下参数
- gene: 差异表达的基因. 不推荐使用"A1BG"这中类型的命名,请用setReadable转换
- OrgDb: 物种注释数据库,一般是org开头
- keytpe: gene的命名格式
- ont: 是BP(Biological Process), CC(Cellular Component), MF(Molecular Function)。一个基因的功能可以从生物学过程,所属细胞部分,和分子功能三个角度定义
只要提供对应的数据即可
ego <- enrichGO( gene = row.names(deseq2.sig), OrgDb = org.hs, keytype = "ENSEMBL", ont = "MF" )
可视化分为冒泡图和网络图等,画起来也就几行代码的事情
dotplot(ego,font.size=5) enrichMap(ego, vertex.label.cex=1.2, layout=igraph::layout.kamada.kawai) plotGOgraph(ego)
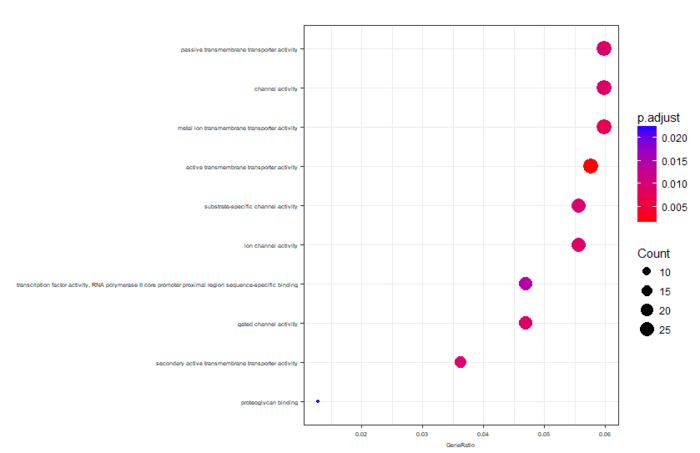
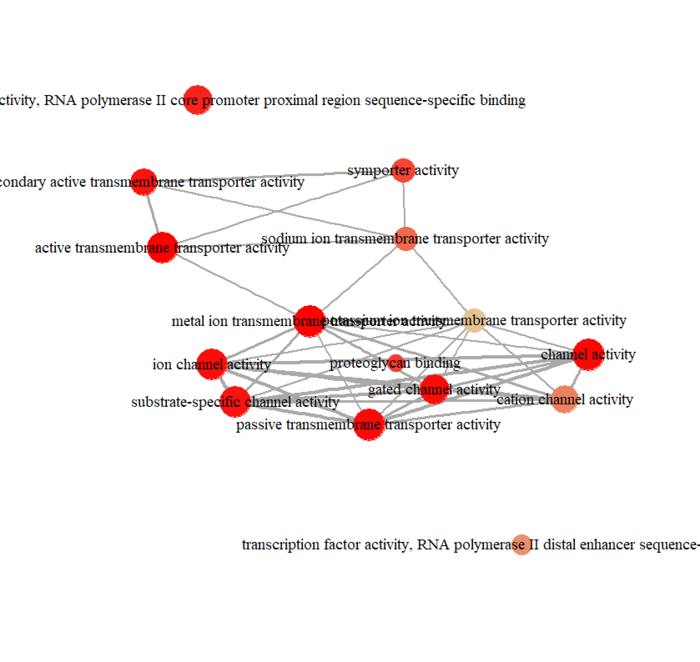
网络图
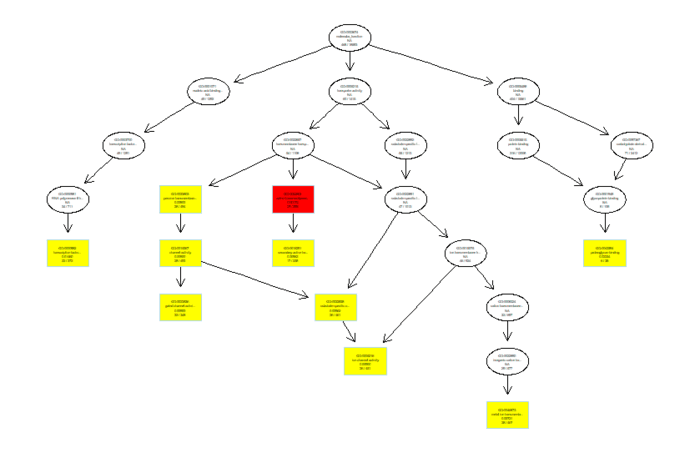
GO图
GSEA分析可使用broadinstitute出品的GSEA可视化软件包
因此我们用clusterProfiler的gseGO或GSEA函数分析,后者可以自定义输入数据
gseGO(geneList, ont = "BP", OrgDb, keyType = "ENTREZID", exponent = 1, nPerm = 1000, minGSSize = 10, maxGSSize = 500, pvalueCutoff = 0.05, pAdjustMethod = "BH", verbose = TRUE, seed = FALSE, by = "fgsea")
geneList: 排序数据, 可以根据log2foldchange, 也可以是pvalues
nPerm: 重抽取次数
minGSSize: 每个基因集的最小数目
maxGSSize: 用于测试的基因注释最大数目
genelist <- sig.deseq2$log2FoldChange
names(genelist) <- rownames(sig.deseq2)
genelist <- sort(genelist, decreasing = TRUE)
gsemf <- gseGO(genelist,
OrgDb = org.hs,
keyType = "ENSEMBL",
ont="MF"
)
head(gsemf)
画一个GSEA标志性图
gseaplot(gsemf, geneSetID="GO:0004871")
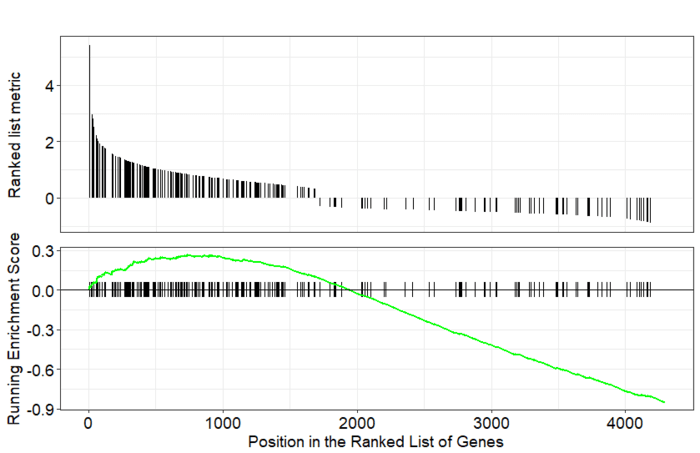
GO:0004871
KEGG富集分析
KEGG富集分析那家强,必须是Y叔的clusterProfiler。因为它能爬取最新的KEGG在线版数据库,而不是用不再更新的KEGG.db。
本部分内容参照KEGG enrichment analysis with latest online data using clusterProfiler
函数是enrichKEGG
enrichKEGG(gene, organism = "hsa", keyType = "kegg", pvalueCutoff = 0.05, pAdjustMethod = "BH", universe, minGSSize = 10, maxGSSize = 500, qvalueCutoff = 0.2, use_internal_data = FALSE)
- gene: 基因名,要和keyType对应
- organism: 需要参考 http://www.genome.jp/kegg/catalog/org_list.html, 人类是hsa
- keyType: 基因的命名方式, "kegg", 'ncbi-geneid', 'ncib-proteinid' and 'uniprot'选择其一
library(clusterProfiler)
gene_list <- mapIds(org.hs, keys = row.names(deseq2.sig),
column = "ENTREZID", keytype = "ENSEMBL" )
kk <- enrichKEGG(gene_list, organism="hsa",
keyType = "ncbi-geneid",
pvalueCutoff=0.05, pAdjustMethod="BH",
qvalueCutoff=0.1)
head(summary(kk))
# 气泡图等图限于篇幅不画了,流量有限
dotplot(kk)注意: 一定要提供符合keyType要求的基因名
总结
你以为结束了?这只是刚开始!我还得修修补补,说不定能出书呢!
参考文献
[1] Guangchuang Yu., Li-Gen Wang, Yanyan Han, Qing-Yu He. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS: A Journal of Integrative Biology. 2012, 16(5):284-287.
[2] Ten Years of Pathway Analysis: Current Approaches and Outstanding Challenges
1F
转录组入门(8):富集分析 学习
来自外部的引用