

大家在用热图软件,例如,pheatmap绘制热图的之后,经常要面临一个问题就是:
图是画出来了,但如果想从图对应到基因,并查询对应基因的表达量就非常麻烦。因为热图聚类功能会打乱样本和基因的顺序,原来的表达量总表已经发生了变化,查询起来就非常麻烦。
尤其当热图中的基因和样本数非常巨大的时候,我们根本看不清热图中的每个格子,再要直接对应回原表格就更加困难。
例如,下图的热图是样本和基因都进行了聚类。
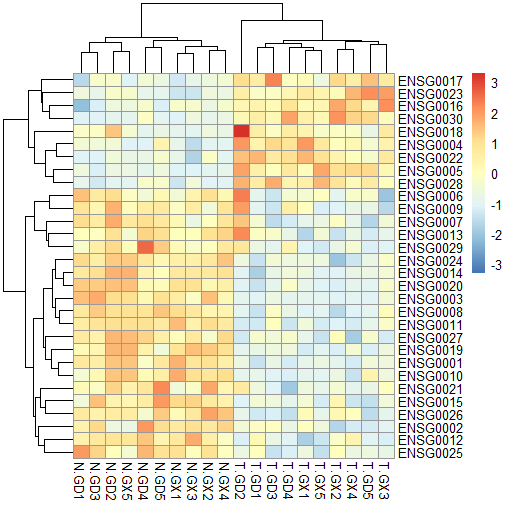
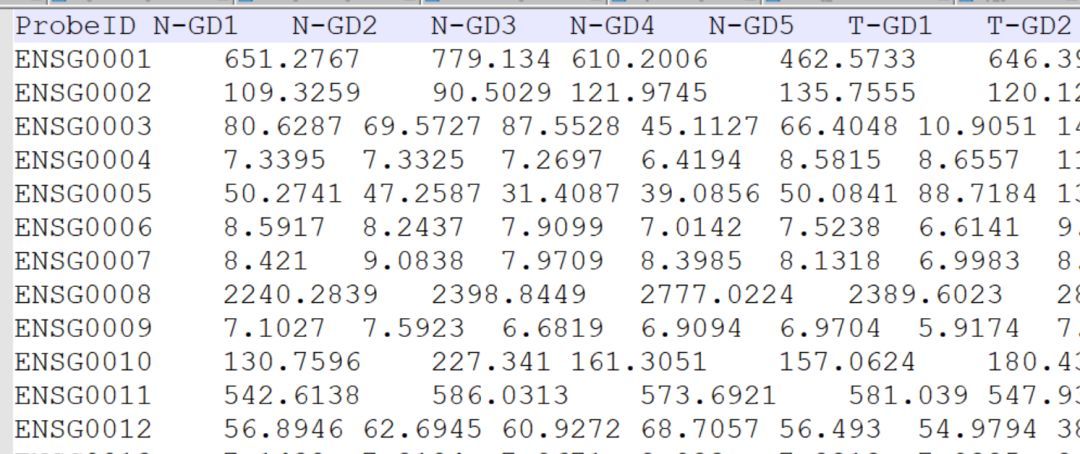
但原绘图文件是这样的,顺序和热图不同。
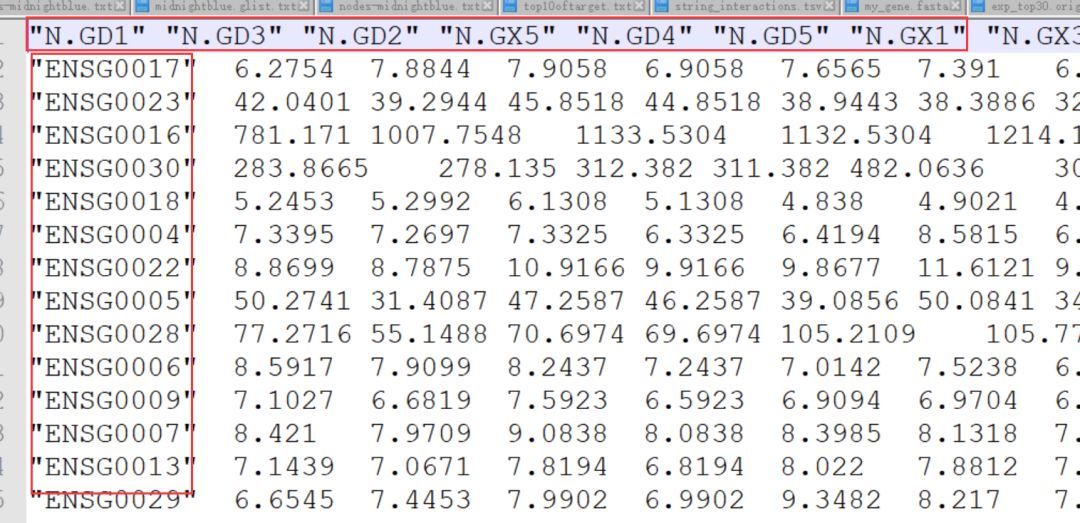
针对这个问题,我们使用R脚本可以轻松解决。以下的R脚本就可以实现输出重排后(与热图保持一致)的表达量表。
重排后的文件这个样子,将和热图顺序保持一致。
#########分割线##########
library(pheatmap)
exp = read.table("exp_top30.original.txt",header=T,row.names=1,sep="t")
result <- pheatmap(exp,scale="row") # 保存热图的绘图关键数据## 从热图中提取样本顺序的编号和样本名
col_oder=result$tree_col$order # 保存热图列顺序(序号)
row_oder=result$tree_row$order # 保存热图行顺序(序号)
cn_new <- colnames(exp)[col_oder] # 保存热图的列名
rn_new <- rownames(exp)[row_oder] # 保存热图的行名
## 生成两个新的与原来的表达量总表长宽相同的数据框(暂时都填写0)
new_exp <- matrix(rep(0,ncol(exp)*nrow(exp)),nrow = nrow(exp),ncol = ncol(exp))
out <- matrix(rep(0,ncol(exp)*nrow(exp)),nrow = nrow(exp),ncol = ncol(exp))
## 将表达量数据读入数据框new_exp,行顺序已经按照热图的顺序重排
for (i in 1:ncol(exp)){
new_exp[,i]=exp[,i][row_oder]
}
## 将表达量数据读入数据框out,列顺序已经进一步按照热图的顺序重排
for (i in 1:ncol(exp)){
out[,i]= new_exp[,col_oder[ i ]]
}
# 将热图的行名和列名导入到排序后的表达量总表中
rownames(out)=rn_new
colnames(out)=cn_new
write.table(out,"hp_exp.txt",sep="t") #输出重排后的表达量表