

这个步骤推荐在R里面做,载入表达矩阵,然后设置好分组信息,统一用DEseq2进行差异分析,当然也可以走走edgeR或者limma的voom流程。
基本任务是得到差异分析结果,进阶任务是比较多个差异分析结果的异同点。
目录
- 数据填坑
- 理论基础:线性模型, 设计矩阵和比较矩阵
- 标准化一二事
- 探索性分析一二事
- 使用DESeq2进行差异基因分析
- 使用edgeR进行差异基因分析
- 使用limma进行差异基因分析
- 不同软件包分析结果比较
- 使用GFOLD进行无重复样本的差异基因分析
- 不同差异表达分析的比较
数据填坑
原先三个样本的HTSeq-count计数的数据可以在我的GitHub中找到,但是前面失误让我们分析的人类就只有3个样本, 另外一个样本需要从另一批数据获取(请注意batch effect),所以不能保证每一组都有两个重复。
我一直坚信”你并不孤独“这几个字,遇到这种情况的人肯定不止我一个,于是我找到了几种解决方法
- 使用edgeR,指定dispersion值
- 无重复转录组数据推荐用同济大学的GFOLD
以上方法都会在后续进行介绍,但是我们DESeq2必须得要有重复的问题亟待解决,没办法我只能自己瞎编了。虽然是编,我们也要有模有样,不能直接复制一份,要考虑到高通量测序的read是默认符合泊松分布的。我是这样编的。
- 计算KD重复组的均值差,作为泊松分布的均值
- 使用概率函数
rpois()随机产生一个数值,前一步的均值作为lambda, - 对一些read count 低于均值的直接加上对应KD重复组之间的差值
# import data if sample are small
options(stringsAsFactors = FALSE)
control <- read.table("F:/Data/RNA-Seq/matrix/SRR3589956.count",
sep="\t", col.names = c("gene_id","control"))
rep1 <- read.table("F:/Data/RNA-Seq/matrix/SRR3589957.count",
sep="\t", col.names = c("gene_id","rep1"))
rep2 <- read.table("F:/Data/RNA-Seq/matrix/SRR3589958.count",
sep="\t",col.names = c("gene_id","rep2"))
# merge data and delete the unuseful row
raw_count <- merge(merge(control, rep1, by="gene_id"), rep2, by="gene_id")
raw_count_filt <- raw_count[-1:-5,]
ENSEMBL <- gsub("(.*?)\\.\\d*?_\\d", "\\1", raw_count_filt$gene_id)
row.names(raw_count_filt) <- ENSEMBL
## the sample problem
delta_mean <- abs(mean(raw_count_filt$rep1) - mean(raw_count_filt$rep2))
sampleNum <- length(raw_count_filt$control)
sampleMean <- mean(raw_count_filt$control)
control2 <- integer(sampleNum)
for (i in 1:sampleNum){
if(raw_count_filt$control[i] < sampleMean){
control2[i] <- raw_count_filt$control[i] + abs(raw_count_filt$rep1[i] - raw_count_filt$rep2[i])
}
else{
control2[i] <- raw_count_filt$control[i] + rpois(1,delta_mean)
}
}
# add data to raw_count
raw_count_filt$control2 <- control2这仅仅是一种填坑的方法而已,更好模拟数据的方法需要参阅更加专业的文献, 有生之年 我希望能补上这一个部分。
理论基础:线性模型, 设计矩阵和比较矩阵
这部分内容最先在 RNA-Seq Data Analysis 的8.5.3节看到,刚开始一点都不理解,但是学完生物统计之后,我认为这是理解所有差异基因表达分析R包的关键。
基本上,统计课都会介绍如何使用t检验用来比较两个样本之间的差异,然后在样本比较多的时候使用方差分析确定样本间是否有差异。当然前是样本来自于正态分布的群体,或者随机独立大量抽样。
对于基因芯片的差异表达分析而言,由于普遍认为其数据是服从正态分布,因此差异表达分析无非就是用t检验和或者方差分析应用到每一个基因上。高通量一次性找的基因多,于是就需要对多重试验进行矫正,控制假阳性。目前在基因芯片的分析用的最多的就是limma。
但是,高通量测序(HTS)的read count普遍认为是服从泊松分布(当然有其他不同意见),不可能直接用正态分布的t检验和方差分析。 当然我们可以简单粗暴的使用对于的非参数检验的方法,但是统计力不够,结果的p值矫正之估计一个差异基因都找不到。老板花了一大笔钱,结果却说没有差异基因,是个负结果,于是好几千经费打了水漂,他肯定是不乐意的。因此,还是得要用参数检验的方法,于是就要说到方差分析和线性模型之间的关系了。
线性回归和方差分析是同一时期发展出的两套方法。在我本科阶段的田间统计学课程中就介绍用方差分析(ANOVA)分析不同肥料处理后的产量差异,实验设计如下
| 肥料 | 重复1 | 重复2 | 重复3 | 重复4 |
|---|---|---|---|---|
| A1 | ... | ... | ... | ... |
| A2 | ... | ... | ... | ... |
| A3 | ... | ... ... | ... | ... |
这是最简单的单因素方差分析,每一个结果都可以看成 yij = ai + u + eij, 其中u是总体均值,ai是每一个处理的差异,eij是随机误差。
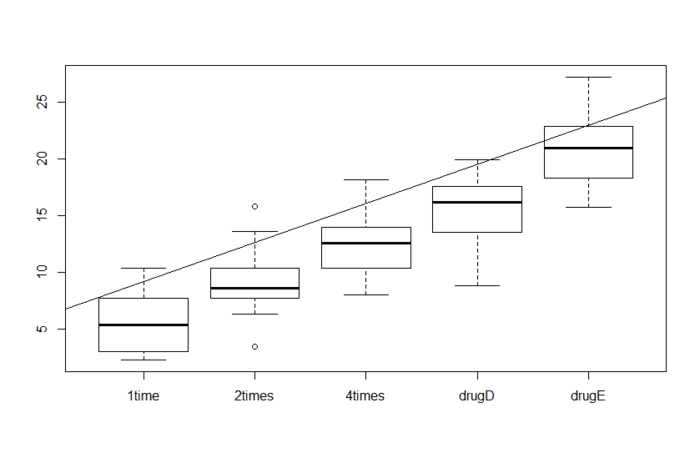
注:方差分析(Analysis of Variance, ANAOVA)名字听起来好像是检验方差,但其实是为了判断样本之间的差异是否真实存在,为此需要证明不同处理内的方差显著性大于不同处理间的方差。
线性回归 一般是用于量化的预测变量来预测量化的响应变量。比如说体重与身高的关系建模:
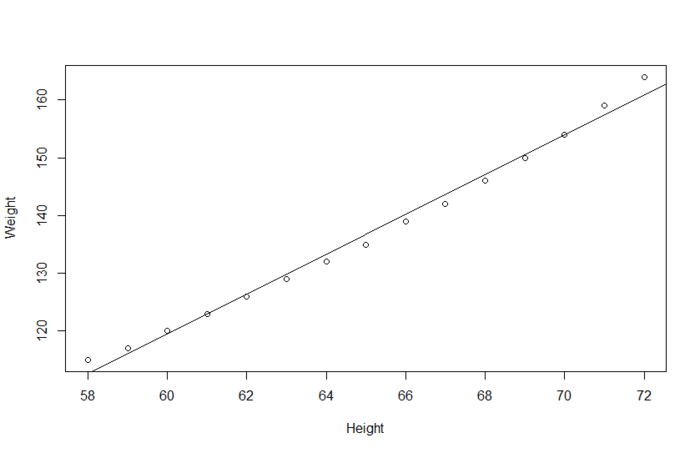
当然线性回归也可用处理名义型或有序型因子(也就是离散变量)作为预测变量,如果要画图的话,就是下面这个情况。

如果我们需要通过一个实验找到不同处理后对照组和控制组的基因变化,那么基因表达可以简单写成, y = a + b · treament + e。 和之前的 yij = ai + u + eij 相比,你会发现公式是如此的一致。 这是因为线性模型和方差分析都是广义线性模型(generalizing linear models, GLM)在正态分布的预测变量的特殊形式。而GLM本身只要采用合适的连接函数是可以处理对任意类型的变量进行建模的。
目前认为read count之间的差异是符合负二项分布,也叫gamma-Possion分布。那么问题来了,如何用GLM或者LM分析两个处理件的差异呢?其实可以简单的用上图的拟合直线的斜率来解释,如果不同处理之间存在差异,那么这个拟合线的斜率必定不为零,也就是与X轴平行。但是这是一种便于理解的方式(虽然你也未必能理解),实际更加复杂,考虑因素更多。
注1 负二向分布有两个参数,均值(mean)和离散值(dispersion). 离散值描述方差偏离均值的程度。泊松分布可以认为是负二向分布的离散值为1,也就是均值等于方差(mean=variance)的情况。
注2 这部分涉及大量的统计学知识,不懂就用维基百科一个个查清楚。
聊完了线性模型和方差分析,下面的设计矩阵(design matrix)就很好理解了, 其实就是用来告诉不同的差异分析函数应该如何对待变量。比如说我们要研究的KD和control之间变化,设计矩阵就是
| 样本 | 处理 |
|---|---|
| sample1 | control |
| sample2 | control |
| sample3 | KD |
| sample4 | KD |
那么比较矩阵(contrast matrix)就是告诉差异分析函数应该如何对哪个因素进行比较, 这里就是比较不同处理下表达量的变化。
标准化一二事
其实read count如何标准化的方法有很多,最常用的是FPKM和RPKM,虽然它们其实是错的--FPKM/RPKM是错的。
我推荐阅读 Comparing the normalization methods for the differential analysis of Illumina high-throughput RNA-Seq data , 了解不同标准化方法之间的差异。
有一些方法是要求原始数据,有一些则要求经过某类标准化后的数据,记得区分。
探索性分析一二事
使用DESeq2进行差异基因分析
关于DESeq2分析差异表达基因,其实在https://www.bioconductor.org/help/workflows/rnaseqGene/ 里面介绍的非常清楚了。
我们已经准备好了count matrix,接下来就是把数据导入DESeq2。DESeq2导入数据的方式有如下4种,基本覆盖了主流read count软件的结果。
注 DESeq2要求的数据是raw count, 没必要进行FPKM/TPM/RPFKM/TMM标准化。
| function | package | framework | output | DESeq2 input function |
|---|---|---|---|---|
| summarizeOverlaps | GenomicAlignments | R/Bioconductor | SummarizedExperiment | DESeqDataSet |
| featureCounts | Rsubread | R/Bioconductor | matrix | DESeqDataSetFromMatrix |
| tximport | tximport | R/Bioconductor | list of matrices | DESeqDataSetFromTximport |
| htseq-count | HTSeq | Python | files | DESeqDataSetFromHTSeq |
本来我们是可以用DESeq2为htseq-count专门提供的 DESeqDataSetFromHTSeq ,然而很尴尬数据不够要自己凑数,所以只能改用 DESeqDataSetFromMatrix了 :cold_sweat:
导入数据,构建 DESeq2 所需的 DESeqDataSet 对象
library(DESeq2)
countData <- raw_count_filt[,2:5]
condition <- factor(c("control","KD","KD","control"))
dds <- DESeqDataSetFromMatrix(countData, DataFrame(condition), design= ~ condition )注: 这一步到下一步之间可以过滤掉一些low count数据,节省内存,提高运行速度
nrow(dds) dds <- dds[ rowSums(counts(dds)) > 1, ] nrow(dds)
使用DESeq进行差异表达分析: DESeq包含三步,estimation of size factors(estimateSizeFactors), estimation of dispersion(estimateDispersons), Negative Binomial GLM fitting and Wald statistics(nbinomWaldTest),可以分布运行,也可用一步到位,最后返回 results可用的DESeqDataSet对象。
dds <- DESeq(dds) # 出现如下提示信息,说明运行成功 estimating size factors estimating dispersions gene-wise dispersion estimates mean-dispersion relationship final dispersion estimates fitting model and testing
用results获取结果: results的参数非常的多,这里不好具体展开 :pensive: 但是你们会自己看的吧
res <- results()
我们可用mcols查看每一项结果的具体含义,比如说log2FoldChange 表示倍数变化取log2结果,还能画个火山图。一般简单粗暴的用2到3倍作为阈值,但是对于低表达的基因,3倍也是噪音,那些高表达的基因,1.1倍都是生物学显著了。更重要的没有考虑到组内变异,没有统计学意义。padj 就是用BH对多重试验进行矫正。
mcols(res, use.names = TRUE)
DataFrame with 6 rows and 2 columns
type description
<character> <character>
baseMean intermediate mean of normalized counts for all samples
log2FoldChange results log2 fold change (MLE): condition KD vs control
lfcSE results standard error: condition KD vs control
stat results Wald statistic: condition KD vs control
pvalue results Wald test p-value: condition KD vs control
padj results BH adjusted p-values用summary看描述性的结果,大致是上调的基因占总体的11%,下调的是7.1%(KD vs control)
summary(res) out of 29469 with nonzero total read count adjusted p-value < 0.1 LFC > 0 (up) : 3154, 11% LFC < 0 (down) : 2095, 7.1% outliers [1] : 0, 0% low counts [2] : 15111, 51% (mean count < 22) [1] see 'cooksCutoff' argument of ?results [2] see 'independentFiltering' argument of ?results
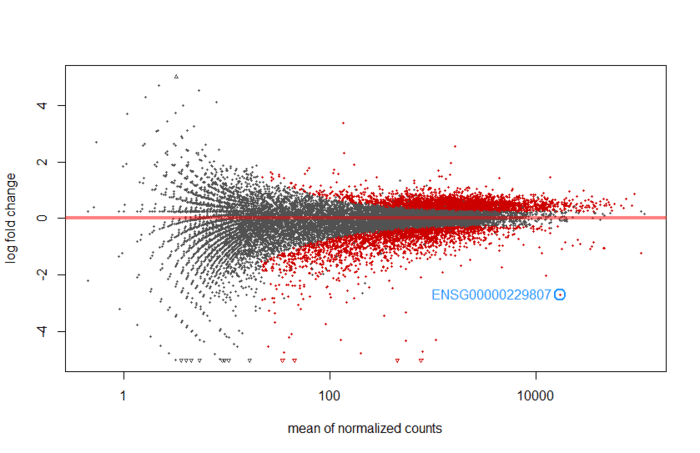
画个MA图,还能标注p值最小的基因。
An MA plot is an application of a Bland–Altman plot for visual representation of genomic data. The plot visualises the differences between measurements taken in two samples, by transforming the data onto M (log ratio) and A (mean average) scales, then plotting these values. Though originally applied in the context of two channel DNA microarray gene expression data, MA plots are also used to visualise high-throughput sequencing analysis --From wikipeida
M表示log fold change,衡量基因表达量变化,上调还是下调。A表示每个基因的count的均值。根据summary可知,low count的比率很高,所以大部分基因表达量不高,也就是集中在0的附近(log2(1)=0,也就是变化1倍).提供了模型预测系数的分布总览。
下图是没有经过 statistical moderation平缓log2 fold changes的情况
plotMA(res, ylim = c(-5,5))
topGene <- rownames(res)[which.min(res$padj)]
with(res[topGene, ], {
points(baseMean, log2FoldChange, col="dodgerblue", cex=2, lwd=2)
text(baseMean, log2FoldChange, topGene, pos=2, col="dodgerblue")
})
如果经过lfcShrink 收缩log2 fold change, 结果会好看很多
res.shrink <- lfcShrink(dds, contrast = c("condition","KD","control"), res=res)
plotMA(res.shrink, ylim = c(-5,5))
topGene <- rownames(res)[which.min(res$padj)]
with(res[topGene, ], {
points(baseMean, log2FoldChange, col="dodgerblue", cex=2, lwd=2)
text(baseMean, log2FoldChange, topGene, pos=2, col="dodgerblue")
})
当然还有火山图,不过留给其他方法作图,我们先把差异表达的基因找出来。
res.deseq2 <- subset(res, padj < 0.05)
一般p value 小于0.05就是显著了, 显著性不代表结果正确,只用于给后续的富集分析和GSEA提供排序标准和筛选而已。关于P值的吐槽简直无数, 请多注意。
使用edgeR进行差异基因分析
edgeR在函数说明中称其不但可以分析SAGE, CAGE的RNA-Seq,Tag-RNA,或RNA-seq, 也能分析ChIP-Seq和CRISPR得到的read counts数据。嗯,我信了:confused:!
edgeR使用DGEList函数读取count matrix数据,也就说你需要提供一个现成的matrix数据,而不是指望它能读取单独的文件,然后进行合并(当然机智的我发现,其实可以用 tximport 或 DESeqDataSetFromHTSeq 读取单独的文件,然后传递给DGEList)
第一步: 构建DGEList对象
library(edgeR)
group <- factor(c("control","KD","KD","control"))
genelist <- DGEList(counts=raw_count_filt[,2:5], group = group)第二步: 过滤 low counts数据。与DESeq2的预过滤不同,DESeq2的预过滤只是为了改善后续运算性能,在运行过程中依旧会自动处理low count数据,edgeR需要在分析前就要排除那些low count数据,而且非常严格。从生物学角度,有生物学意义的基因的表达量必须高于某一个阈值。从统计学角度上, low count的数据不太可能有显著性差异,而且在多重试验矫正阶段还会拖后腿。 综上所诉,放心大胆的过滤吧。
根据经验(又是经验 :dog: ), 基因至少在某一些文库的count超过10 ~ 15 才被认为是表达。这一步全靠尝试, 剔除太多就缓缓,剔除太少就严格点。 我们可以简单的对每个基因的raw count进行比较,但是建议用CPM(count-per-million)标准化 后再比较,避免了文库大小的影响。
# 简单粗暴的方法 keep <- rowSums(genelist$count) > 50 # 利用CPM标准化 keep <- rowSums(cpm(genelist) > 0.5 ) >=2 table(keep) genelist.filted <- genelist[keep, ,keep.lib.sizes=FALSE]
这里的0.5(即阈值)等于 10/(最小的文库的 read count数 /1000000),keep.lib.size=FALSE表示重新计算文库大小。
第三步: 根据组成偏好(composition bias)标准化。edgeR的calcNormFactors函数使用TMM算法对DGEList标准化
genelist.norm <- calcNormFactors(genelist.filted)
注 大部分的mRNA-Seq数据分析用TMM标准化就行了,但是也有例外,比如说single-cell RNA-Seq(Lun, Bach, and Marioni 2016), 还有就是global differential expression, 基因组一半以上的基因都是差异表达的,请尽力避免,(D. Wu et al. 2013), 不然就需要用到内参进行标准化了(Risso et al. 2014).
第四步: 实验设计矩阵(Design matrix), 类似于DESeq2中的design参数。 edgeR的线性模型和差异表达分析需要定义一个实验设计矩阵。很直白的就能发现是1vs0
design <- model.matrix(~0+group) colnames(design) <- levels(group) design control KD 1 1 0 2 0 1 3 0 1 4 1 0
第五步: 估计离散值(Dispersion)。前面已经提到负二项分布(negative binomial,NB)需要均值和离散值两个参数。edgeR对每个基因都估测一个经验贝叶斯稳健离散值(mpirical Bayes moderated dispersion),还有一个公共离散值(common dispersion,所有基因的经验贝叶斯稳健离散值的均值)以及一个趋势离散值
genelist.Disp <- estimateDisp(genelist.norm, design, robust = TRUE) plotBCV(genelist.Disp)
还可以进一步通过quasi-likelihood (QL)拟合NB模型,用于解释生物学和技术性导致的基因特异性变异 (Lund et al. 2012; Lun, Chen, and Smyth 2016).
fit <- glmQLFit(genelist.Disp, design, robust=TRUE) head(fit$coefficients)
注1 估计离散值这个步骤其实有许多estimate*Disp函数。当不存在实验设计矩阵(design matrix)的时候,estimateDisp 等价于 estimateCommonDisp 和 estimateTagwiseDisp 。而当给定实验设计矩阵(design matrix)时, estimateDisp 等价于 estimateGLMCommonDisp, estimateGLMTrendedDisp 和 estimateGLMTagwiseDisp。 其中tag与gene同义。
注2 其实这里的第三, 四, 五步对应的就是DESeq2的DESeq包含的2步,标准化和离散值估测。
第六步: 差异表达检验(1)。这一步主要构建比较矩阵,类似于DESeq2中的results函数的 contrast 参数。
cntr.vs.KD <- makeContrasts(control-KD, levels=design) res <- glmQLFTest(fit, contrast=cntr.vs.KD) ig.edger <- res$table[p.adjust(res$table$PValue, method = "BH") < 0.01, ]
这里用的是glmQLFTest而不是glmLRT是因为前面用了glmQLTFit进行拟合,所以需要用QL F-test进行检验。如果前面用的是glmFit,那么对应的就是glmLRT. 作者称QL F-test更加严格。多重试验矫正用的也是BH方法。
后续就是提取显著性差异的基因用作下游分析,做一些图看看
topTags(res,n=10)
is.de <- decideTestsDGE(res)
summary(is.de)
plotMD(res, status=is.de, values=c(1,-1), col=c("red","blue"),
legend="topright")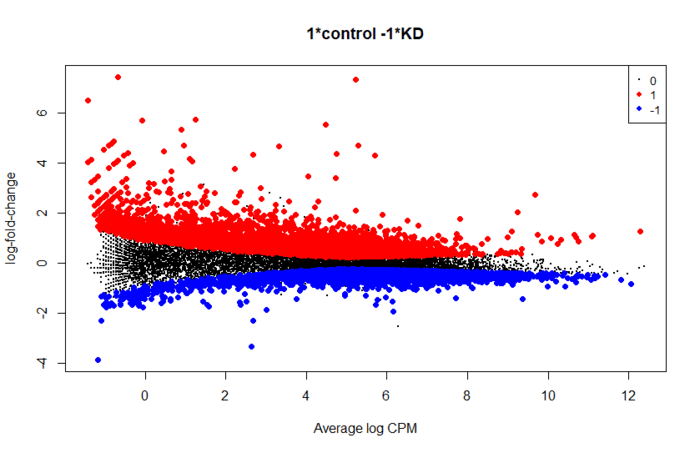
第六步:差异表达检验(2)。上面找到的显著性差异的基因,没有考虑效应值,也就是具体变化了多少倍。我们也可用找表达量变化比较大的基因,对应的函数是 glmTreat。
tr <- glmTreat(fit, contrast=B.LvsP, lfc=log2(1.5)) s
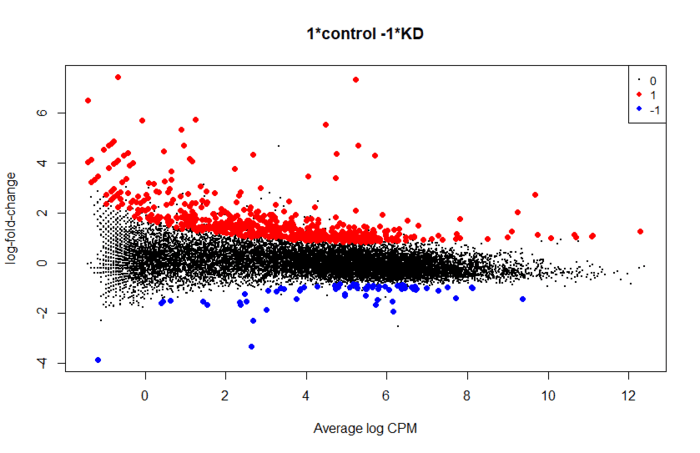
使用limma进行差异分析
经过上面两个方法的洗礼,基本上套路你也就知道了,我先简单小结一下,然后继续介绍limma包的 voom 。
- 导入read count, 保存为专门的对象用于后续分析
- 原始数据过滤,根据标准化read count 或者 raw count 作为筛选标准
- raw read count 标准化
- 通过各种算法(如经验贝叶斯,EM)预测dispersion离散值
- 广义线性模型拟合数据
- 差异分析,也就是统计检验部分
Limma原先用于处理基因表达芯片数据,可是说是这个领域的老大 :sunglasses: 。如果你仔细看edgeR导入界面,你就会发现,edgeR有一部分功能依赖于limma包。Limma采用经验贝叶斯模型( Empirical Bayesian model)让结果更稳健。
在处理RNA-Seq数据时,raw read count先被转成log2-counts-per-million (logCPM),然后对mean-variance关系建模。建模有两种方法:
- 精确权重法(precision weights)也就是“voom"
- 经验贝叶斯先验趋势(empirical Bayes prior trend),也就是”limma-trend“
数据预处理: Limma使用edgeR的DGEList对象,并且过滤方法都是一致的,对应edgeR的第一步,第二步, 第三步
library(edgeR)
library(limma)
group <- factor(c("control","KD","KD","control"))
genelist <- DGEList(counts=raw_count_filt[,2:5], group = group)
### filter base use CPM
keep <- rowSums(cpm(genelist) > 0.5 ) >=2
table(keep)
genelist.filted <- genelist[keep, ,keep.lib.sizes=FALSE]
### normalizaition
x <- calcNormFactors(x, method = "TMM")差异表达分析: 使用”limma-trend“
design <- model.matrix(~0+group) colnames(design) <- levels(group) logCPM <- cpm(genelist.norm, log=TRUE, prior.count=3) fit <- lmFit(logCPM, design) fit <- eBayes(fit, trend=TRUE) topTable(fit, coef=ncol(design))
差异表达分析: 使用”limma-voom“
### DGE with voom v <- voom(genelist.norm, design, plot=TRUE) #v <- voom(counts, design, plot=TRUE) fit <- lmFit(v, design) fit <- eBayes(fit) all <- topTable(fit, coef=ncol(design), number=10000) sig.limma <- all[all$adj.P.Val < 0.01, ] fit <- treat(fit, lfc=log2(1.2)) topTreat(fit, coef=ncol(design))
如果分析基因芯片数据,必须好好读懂LIMMA包。
不同软件包分析结果比较
基本上每一个包,我都提取了各种的显著性基因,比较就需要用韦恩图了,但是我偏不 :stuck_out_tongue: 我要用UpSetR.
library(UpSetR) input <- fromList(list(edgeR=rownames(sig.edger), DESeq2=rownames(sig.deseq2), limma=rownames(sig.limma)))
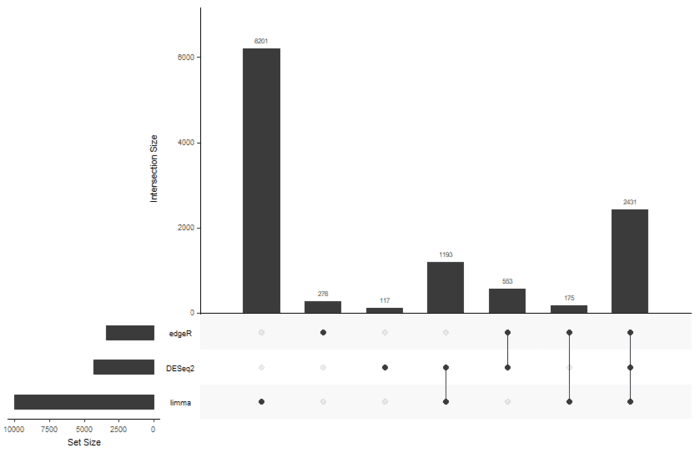
感觉limma的结果有点奇怪,有生之年在折腾吧。
使用GFOLD进行无重复样本的差异基因分析
参考文件
[1] Comparing the normalization methods for the differential analysis of Illumina high-throughput RNA-Seq data
[2] https://www.bioconductor.org/help/workflows/rnaseqGene/
[3] https://www.bioconductor.org/help/workflows/RnaSeqGeneEdgeRQL/
1F
转录组入门(7):差异表达分析 学习