

本文来自百迈客基因微信公众号(BMK_product)
随着测序成本的不断降低,转录组测序分析已逐渐成为一种很常用的分析手段。但对于转录组分析当中的一些概念,很多人还不是很清楚。今天,小编就来谈谈在转录组分析中,经常会遇到的一个概念FDR,那什么是FDR?为什么要用FDR呢?一起来学习吧!
什么是FDR
FDR (false discovery rate),中文一般译作错误发现率。在转录组分析中,主要用在差异表达基因的分析中,控制最终分析结果中,假阳性结果的比例。
为什么要用FDR
在转录组分析中,如何确定某个转录本在不同的样品中表达量是否有差异是分析的核心内容之一。一般来说,我们认为,不同样品中,表达量差异在两倍以上的转录本,是具有表达差异的转录本。为了判断两个样品之间的表达量差异究竟是由于各种误差导致的还是本质差异,我们需要根据所有基因在这两个样本中的表达量数据进行假设检验。常用的假设检验方法有t-检验、卡方检验等。很多刚接触转录组分析的人可能会有这样一个疑问,一个转录本是不是差异表达,做完假设检验看P-value不就可以了么?为什么会有FDR这样一个新的概念出现?这是因为转录组分析并不是针对一个或几个转录本进行分析,转录组分析的是一个样品中所转录表达的所有转录本。所以,一个样品当中有多少转录本,就需要对多少转录本进行假设检验。这会导致一个很严重的问题,在单次假设检验中较低的假阳性比例会累积到一个非常惊人的程度。举个不太严谨的例子。
假设现在有这样一个项目:
● 包含两个样品,共得到10000条转录本的表达量数据,
● 其中有100条转录本的表达量在两个样品中是有差异的。
● 针对单个基因的差异表达分析有1%的假阳性。
由于存在1%假阳性的结果,在我们分析完这10000个基因后,我们会得到100个假阳性导致的错误结果,加上100条真实存在的结果,共计200个结果。在这个例子中,一次分析得到的200个差异表达基因中,有50%都是假阳性导致的错误结果,这显然是不可接受的。为了解决这个问题,FDR这个概念被引入,以控制最终得到的分析结果中假阳性的比例。
如何计算FDR
FDR的计算是根据假设检验的P-value进行校正而得到的。一般来说,FDR的计算采用Benjamini-Hochberg方法(简称BH法),计算方法如下:
1. 将所有P-value升序排列.P-value记为P,P-value的序号记为i,P-value的总数记为m
2. FDR(i)=P(i)*m/i
3. 根据i的取值从大到小,依次执行FDR(i)=min{FDR(i),FDR(i+1)}
注:实际上,BH法的原始算法是找到一个最大的i,满足P≤i/m*FDR阈值,此时,所有小于i的数据就都可以认为是显著的。在实践中,为了能够在比较方便的用不同的FDR阈值对数据进行分析,采用了步骤3里的方法。这个方法可以保证,不论FDR阈值选择多少,都可以直接根据FDR的数值来直接找到所有显著的数据。
下面我们以一个包含10个数据的例子来看一下FDR计算的过程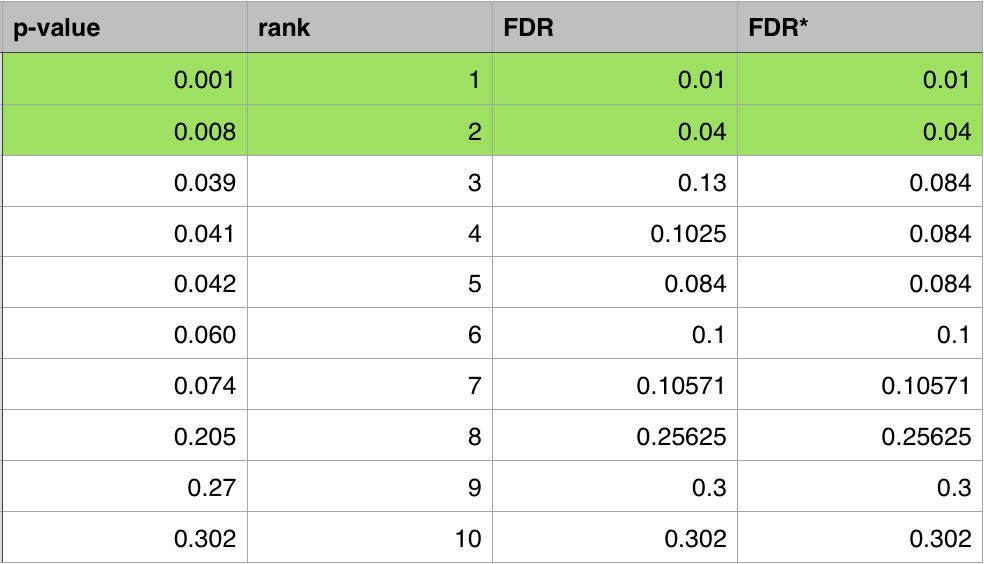
在这个例子中,第一列是原始的P-value,第二列是排序后的序号,第三列是根据P-value校正得到的初始FDR,第四列是最终用于筛选数据的FDR数值。如果我们设定FDR<0.05,那么绿色高亮的两个数据就是最终分析认为显著的数据。
FDR的阈值选择在转录组分析中是非常重要的一个环节,常用的阈值包括0.01、0.05、0.1等。实践中也可以根据实际的需要来灵活选择。例如,在做真菌或者原核生物的转录组分析时,由于这些物种转录本数量较少,假阳性累积的程度较低,所以可以适当将FDR阈值设置的较高一些,这样可以获得较多的差异表达结果,有利于后续的分析。