

首先当然是下载软件啦!
两个地方可以下载,一个是谷歌code中心,被墙啦,另一个是github,我的最爱。
wget https://codeload.github.com/alexdobin/STAR/zip/master
解压即可使用啦,其中程序在bin目录下面,根据自己的平台调用即可!
然后doc里面还有个pdf的说明文档,写的非常清楚,我也是看着那个文档学的这个软件!
接下来就是准备数据啦!
既然是类似于tophat一样的比对软件,当然是准备参考基因组和测序数据咯,毫无悬念。
然后 该软件也给出了一些测试数据
ftp://ftp2.cshl.edu/gingeraslab/tracks/STARrelease/2.1.4/
然后就是运行程序的命令!
分为两个步骤:首先构建索引,然后比对即可,中间的参数根据具体需要可以细调!
构建索引时候,软件说明书给的例子是
The basic options to generate genome indices are as follows:
–runThreadN NumberOfThreads
–runMode genomeGenerate
–genomeDir /path/to/genomeDir
–genomeFastaFiles /path/to/genome/fasta1 /path/to/genome/fasta2 …
–sjdbGTFfile /path/to/annotations.gtf
–sjdbOverhang ReadLength-1
我模仿了一下。对我从ensembl ftp里面下载的老鼠基因组构建了索引
/home/jmzeng/hoston/RNA-soft/STAR-master/bin/Linux_x86_64/STAR \ –runThreadN 30 #我的服务器还比较大,可以使用30个CPU \ –runMode genomeGenerate \ –genomeDir /home/jmzeng/hoston/mouse/STAR-mouse #构建好的索引放在这个目录 \ –genomeFastaFiles /home/jmzeng/hoston/mouse/Mus_musculus.GRCm38.fa.fa \ –sjdbGTFfile /home/jmzeng/hoston/mouse/Mus_musculus.GRCm38.79.gtf \ –sjdbOverhang 284 #我的测序数据长短不一,最长的是285bp
当然注释的地方你要删除掉才行呀,因为cpu用的比较多。
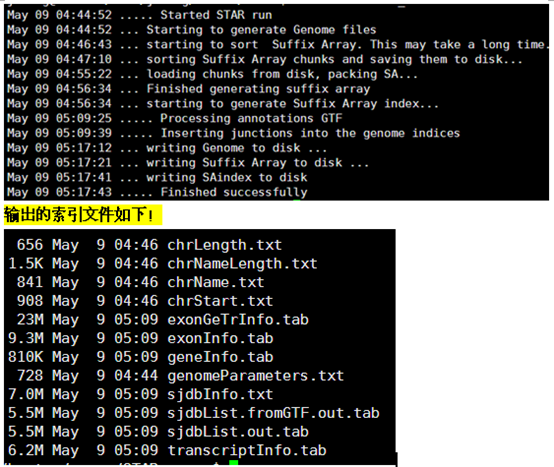
算一算时间,对4.6G的小鼠基因组来说,半个小时算是非常快的了!Bowtie2的index要搞两个多小时。
然后就是比对咯。这也是很简单的,软件说明书给的例子是
The basic options to run a mapping job are as follows:
–runThreadN NumberOfThreads
–genomeDir /path/to/genomeDir
–readFilesIn /path/to/read1 [/path/to/read2]
我稍微理解了一下参数,然后写出了自己的命令。
fq=740WT1.fq.trimmed.single mkdir 740WT1_star /home/jmzeng/hoston/RNA-soft/STAR-master/bin/Linux_x86_64/STAR \ –runThreadN 20 \ –genomeDir /home/jmzeng/hoston/mouse/STAR-mouse \ –readFilesIn $fq \ –outFileNamePrefix ./740WT1_star/740WT1
如果输出文件需要被cufflinks套装软件继续使用。就需要用一下参数
Cufflinks/Cuffdiff require spliced alignments with XS strand attribute, which STAR will generate with –outSAMstrandField intronMotif option.
还有–outSAMtype参数可以修改输出比对文件格式,可以是sam也可以是bam,可以是sort好的,也可以是不sort的。
最后是输出文件解读咯!
其实没什么好解读的,输出反正就是sam类似的比对文件咯,如果还有其它文件,需要自己好好解读说明书啦。我就不废话了!
值得一提的是,该程序提供了2次map的建议
The basic idea is to run 1st pass of STAR mapping with the usual parameters , then collect the junctions detected in the first pass, and use them as ”annotated” junctions for the 2nd pass mapping.
在对RNA-seq做snp-calling的时候可以用到,尤其是GATK官方还给出了教程,大家可以好好学习学习!
http://www.broadinstitute.org/gatk/guide/article?id=3891
原文来自:http://www.bio-info-trainee.com/727.html