

StringTie由约翰霍普金斯大学联合德州大学西南医学中心开发,能够组装转录本并预计表达水平。它应用网络流算法和可选的denovo组装,将复杂的数据集组装成转录本。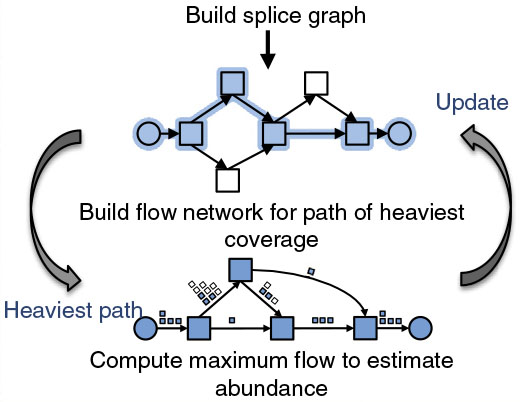
相对于其他拼接软件(Cufflinks, IsoLasso, Scripture,Traph等),StringTie能够拼接出更完整、更准确的基因,并且StringTie采用拼接和定量同步进行,相对于其他方法,其定量结果更加准确。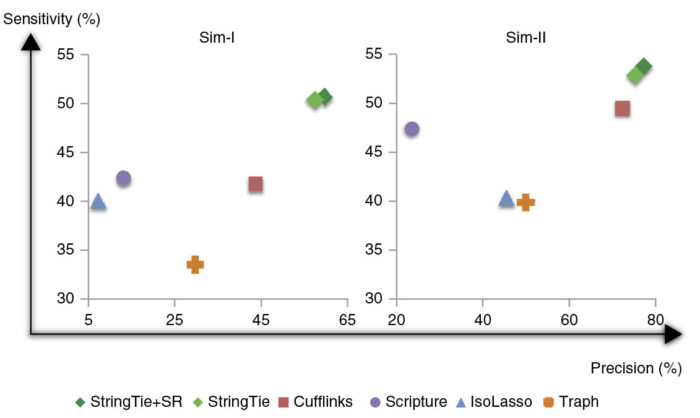
文中指出,对于从人类血液中获得的reads,StringTie正确组装了10,990个转录本,而Cufflinks只组装了7,187个。而对于模拟的数据集,StringTie正确组装了7,559个转录本,比Cufflinks的6,310个提高了20%。此外,它的运行速度也比其他组装软件更快。
软件安装
下载StringTie并解压:
tar zxvf stringtie-1.1.2.Linux_x86_64.tar.gz
将HISAT2目录添加到环境变量:
vi ~/.bashrc
在文件末位添加:
export PATH=/home/chenwen/bin/stringtie-1.1.2.Linux_x86_64:$PATH
保存退出
source ~/.bashrc
StringTie的输入文件
HISAT的输出文件为SAM格式,需要经过两步转换成StringTie可以使用的BAM格式:
1. SAM转BAM,并排序:
samtools view -S -b input.sam | samtools sort – input.sorted
2.修改HI标签:
samtools view -h input.bam | perl -ne ‘if(/HI:i:(d )/) {$m=$1-1; $_ =~ s/HI:i:(d )/HI:i:$m/} print $_;’| samtools view -bS – > input.correct.bam
运行StringTie
stringtie SRR534294.correct.bam -p 16 -G genes.gtf -B -o stout/transcripts.gtf
-p 指定线程数,默认1
-G 指定参考的转录组注释文件
-B 生成用于Ballgown 分析的文件
-o 指定输出文件
更多参数请查看HISAT2的操作手册:
https://ccb.jhu.edu/software/stringtie/index.shtml?t=manual