

大家都知道,这十几年来最流行的差异分析软件就是R的limma包了,但是它以前只支持microarray的表达数据。
考虑到大家都熟悉了它,它又发了一个voom的方法,让它从此支持RNA-seq的count数据啦!大家都知道芯片数据跟RNA-seq数据的本质就是value的分布不一样,所以各种针对RNA-seq的差异分析包也就是提出来一个新的normalization方法而已。而我们limma本身就提出了一个voom的方法来对RNA-seq数据进行normalization
使用这个包也需要三个数据:
- 表达矩阵
- 分组矩阵
- 差异比较矩阵
用法与limma一模一样的,只是多了一个normalization而已。
读取自己的数据
一般我们会自己读取表达矩阵和分组信息,下面是一个例子:
options(warn=-1) suppressMessages(library(DESeq2)) suppressMessages(library(limma)) suppressMessages(library(pasilla)) data(pasillaGenes) exprSet=counts(pasillaGenes) head(exprSet) ##表达矩阵如下
## treated1fb treated2fb treated3fb untreated1fb untreated2fb ## FBgn0000003 0 0 1 0 0 ## FBgn0000008 78 46 43 47 89 ## FBgn0000014 2 0 0 0 0 ## FBgn0000015 1 0 1 0 1 ## FBgn0000017 3187 1672 1859 2445 4615 ## FBgn0000018 369 150 176 288 383 ## untreated3fb untreated4fb ## FBgn0000003 0 0 ## FBgn0000008 53 27 ## FBgn0000014 1 0 ## FBgn0000015 1 2 ## FBgn0000017 2063 1711 ## FBgn0000018 135 174
group_list=pasillaGenes$condition
## [1] treated treated treated untreated untreated untreated untreated ## Levels: treated untreated ##这是分组信息,7个样本,3个处理的,4个未处理的对照!
另外一个例子是从airway里面得到表达矩阵和分组信息
library(airway) data(airway) exprSet=assays(airway)$counts ## 表达矩阵 group_list=colData(airway)$dex ## 分组信息
第一步:构建分组矩阵
suppressMessages(library(limma)) design <- model.matrix(~factor(group_list)) colnames(design)=levels(factor(group_list)) rownames(design)=colnames(exprSet) design
## treated untreated ## treated1fb 1 0 ## treated2fb 1 0 ## treated3fb 1 0 ## untreated1fb 1 1 ## untreated2fb 1 1 ## untreated3fb 1 1 ## untreated4fb 1 1 ## attr(,"assign") ## [1] 0 1 ## attr(,"contrasts") ## attr(,"contrasts")$`factor(group_list)` ## [1] "contr.treatment"
第二步:根据分组信息和表达矩阵进行normalization
这里构建了一个对象 An object of class “EList”
v <- voom(exprSet,design,normalize="quantile")
## 下面的代码如果你不感兴趣不需要运行,免得误导你
## 就是看看normalization前面的数据分布差异
#png("RAWvsNORM.png")
exprSet_new=v$E
par(cex = 0.7)
n.sample=ncol(exprSet)
if(n.sample>40) par(cex = 0.5)
cols <- rainbow(n.sample*1.2)
par(mfrow=c(2,2))
boxplot(exprSet, col = cols,main="expression value",las=2)
boxplot(exprSet_new, col = cols,main="expression value",las=2)
hist(exprSet)
hist(exprSet_new)
#dev.off()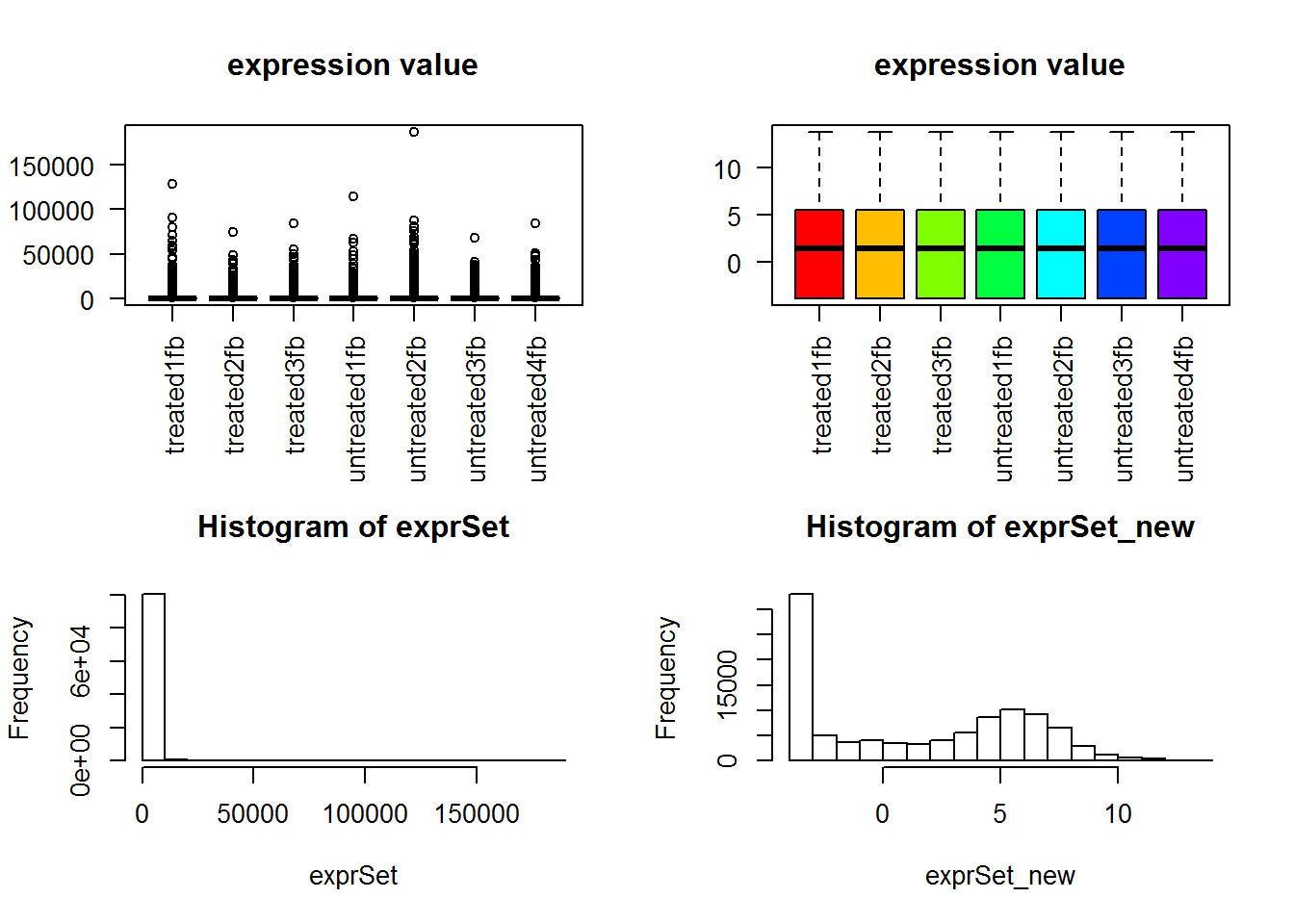
可以很明显看到normalization前后数据分布差异非常大!
第三步:做差异分析,提取差异分析结果
这里不需要差异比较矩阵了,因为我的分组矩阵表明样本就分成两组,直接提取coef=2的数据即可
fit <- lmFit(v,design) fit2 <- eBayes(fit) tempOutput = topTable(fit2, coef=2, n=Inf) DEG_voom = na.omit(tempOutput) head(DEG_voom)
## logFC AveExpr t P.Value adj.P.Val ## FBgn0029167 2.1608689 7.880589 41.19142 5.195806e-08 0.0007518331 ## FBgn0003137 -0.9560776 8.421903 -29.97211 2.920473e-07 0.0021129620 ## FBgn0003748 -1.0385933 6.395990 -23.78787 1.020682e-06 0.0049230892 ## FBgn0035085 2.5190255 5.221183 21.01339 1.993995e-06 0.0058809216 ## FBgn0050185 -0.4886279 5.487547 -19.95422 2.635106e-06 0.0058809216 ## FBgn0015568 -1.1040979 3.922438 -19.96954 2.624231e-06 0.0058809216 ## B ## FBgn0029167 9.065020 ## FBgn0003137 7.800063 ## FBgn0003748 6.652695 ## FBgn0035085 5.870585 ## FBgn0050185 5.734162 ## FBgn0015568 5.55756
差异分析结果就不解释啦!
原文来自:www.bio-info-trainee.com