

bedtools multicov和htseq-count都可以用来对基因和转录本的表达量的计算!!!
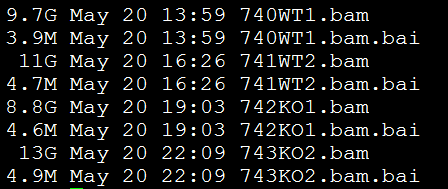
我们总共有四个样本,已经比对到小鼠的mm9基因组上面了,数据大小如下
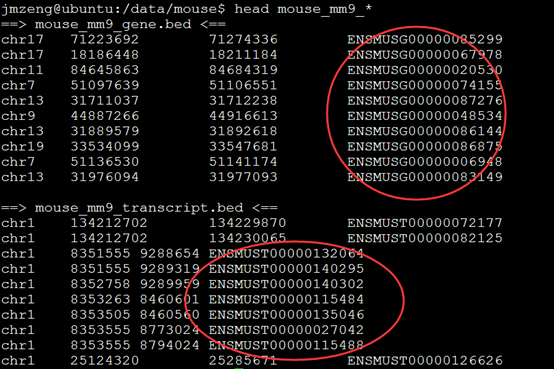
然后对基因和转录本计数需要一些额外的信息,即各个基因及转录本的位置信息,gtf文件需要在UCSC等各大数据库下载
然后我们制作一个config文件配置我们的数据地址
cat sample_bam.config #可以看到文件内容如下 /data/mouse/ptan/740WT1.bam /data/mouse/ptan/741WT2.bam /data/mouse/ptan/742KO1.bam /data/mouse/ptan/743KO2.bam
几个批处理文件名及内容分别如下
bedtools_multicov.sh bedtools_multicov_transcript.sh htseq.sh htseq_transcript.sh while read id do echo $id new=`echo $id |cut -d”/” -f 5` echo $new bedtools multicov -bams $id -bed /data/mouse/mouse_mm9_gene.bed > $new.gene.bedtools_multicov.count done <$1 while read id do echo $id new=`echo $id |cut -d”/” -f 5` echo $new bedtools multicov -bams $id -bed /data/mouse/mouse_mm9_transcript.bed > $new.transcript.bedtools_multicov.count done <$1 while read id do echo $id new=`echo $id |cut -d”/” -f 5` htseq-count -f bam $id /data/mouse/Mus_musculus.NCBIM37.67.gtf.chr > $new.gene.htseq.count done <$1 while read id do echo $id new=`echo $id |cut -d”/” -f 5` htseq-count -f bam –idattr transcript_id $id /data/mouse/Mus_musculus.NCBIM37.67.gtf.chr > $new.transcript.htseq.count done <$1
批量运行这些程序后就能对它们分别分情况进行计数,也能比较这两种计数方法的区别!
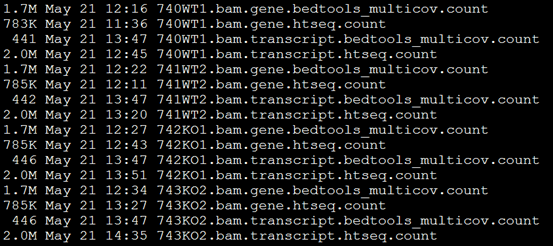
可以看出区别还是很大的!!!
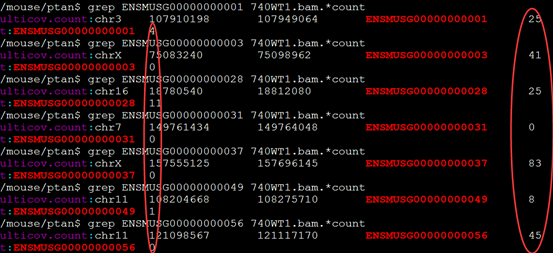
我肯定没搞懂它们的原理,这完全就不一样,已经不是区别的问题了!!!
对于每个个体输出的计数文件,接下来就可以用DESeq等包来进行差异基因分析啦!
原文来自:http://www.bio-info-trainee.com/755.html