

本系列课程学习的文章是:AKAP95 regulates splicing through scaffolding RNAs and RNA processing factors. Nat Commun 2016 Nov 8;7:13347. PMID: 27824034
很容易在文章里面找到数据地址GSE81916 这样就可以下载sra文件
数据下载部分
第一步:在PubMeb上查找文献

第二步: 根据文献的method部分找到RNA-Seq是如何存放的

第三步: 在GEO上查找GSE81916
GEO站点: https://www.ncbi.nlm.nih.gov/geo/
找到了NCBI的SRA工具下载所需要的SRR编号。

GEO网址: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE81916 分为两个部分:
- 共同部分:https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=
- 变动部分:GSE81916
FTP网址ftp://ftp-trace.ncbi.nlm.nih.gov/sra/sra-instant/reads/ByStudy/sra/SRP/SRP075/SRP075747 可以分为以下几个部分
- 所有SRA数据的共同部分: ftp://ftp-trace.ncbi.nlm.nih.gov/sra/sra-instant
- reads表示存放reads数据,在FTP可以看到另一个选项是analysis,表示分析结果
- ByStudy表示根据Study进行分类,其他还可以根据实验
ByExp,根据Run,ByRun. - sra/SRP/SRP075/SRP075747: 后面部分都是为了便于检索。
第四步:通过循环,分别用prefetch下载数据
for i in `seq 48 62`;
do
prefetch SRR35899${i}
done
知识点:如何用循环批量下载数据
注: 数据很大,需要下载很久,这段时间去看文章所用的分析方法。
文章所用方法:
内容主要在Bioinformatic analyses部分
比对:
- 比对软件:TopHat (v2.0.13)
- 参考基因组:human reference genome (GRCh37/hg19)
- GTF文件: GTF version GRCh37.70
- 只保留MQ >30的map结果
- Picard-tools (v1.126): 计算平均插入大小(mean insert sizes)和标准差
read count: 软件:HTSeq v0.6.0
差异表达分析: DESeq (v3.0)
差异外显子使用分析: DEXSeq (v3.1)
GO富集分析:DAVID (http://david.ncifcrf.gov/).
实验设计:
样本9-15为mRNA-Seq测序结果,用于分析人类293个细胞(9-11)和小鼠ES细胞(12-15)d的AKAP95敲出影响。
文章到底用RNA-Seq做了那些事情
为了评估AKAP95对AS的全局影响,他们删除了人类293 cell和小鼠ES细胞,通过RNA-Seq和DEXseq 分析找到细胞mRNA的不同外显子使用。由于DEXseq考虑到了生物学变异,因此对假阳性(False discovery)有可信的控制。在 293 cell 和 ES cell中,AKAPP95 KD都导致更多地外显子使用减少,意味着APAP95通过促进外显子融合调节全局地可变剪切(AS). 他们用PCR-based assay验证了结果。
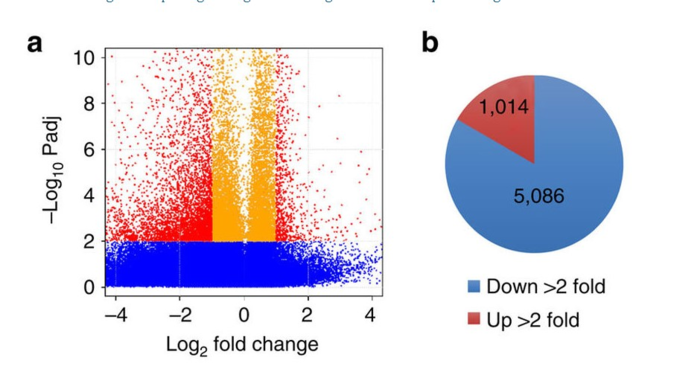
文章用了火山图展示被影响地外显子,用饼图可视化多少个外显子被下调了。Fold change is the ratio of the normalized exon level in AKAP95 KD over that in control cells.
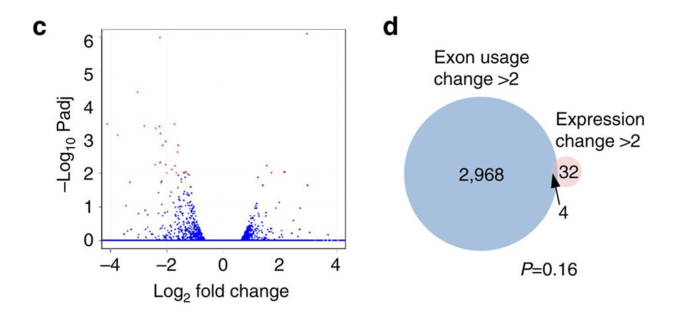
为了证明外显子使用(exon usage)降低不是因为基因表达量降低导致的技术偏差,作者从三个角度进行论证
- 工具角度,DEXseq根据基因的总外显子信号水平标准化每个外显子信号
- 数据分析,AKAP95 KD的细胞中那些外显子使用被影响的大部分基因,表达量没有降低,所以和表达量无关,还用图证明。Fold change is the ratio of the normalized exon level in AKAP95 KD over that in control cells.
- PCR数据证实
- 小鼠的也是如此
确定可变外显子使用是AKAP95的直接影响, 他们比较了AKAP95物理靶点(基于AKAP95 RIP-Seq)和功能位点(基于mRNA-Seq)。 那些AKAP95结合到内含子的基因和外显子使用显著性变化(AKAP95 KD)的基因显著性重叠。
逻辑就是: 如果A和B有关,那么有A就有B, 没有A就没有B,且这种关系不是偶然的。
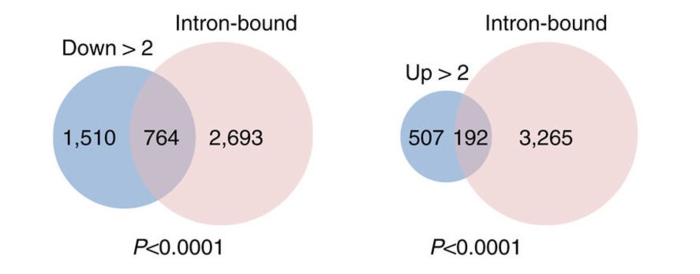
确定AKAP95靶点参与的生物学通路,他们用了基因本体论(GO)分析了AKAP95的功能位点和物理位点。结果揭示那些AKAP95 KD 的293细胞中那些差异外显子使用的基因,显著性的富集在chromatin/transcription regulators and RNA processing factors。那些RIP-Seq找到基因也是如此。
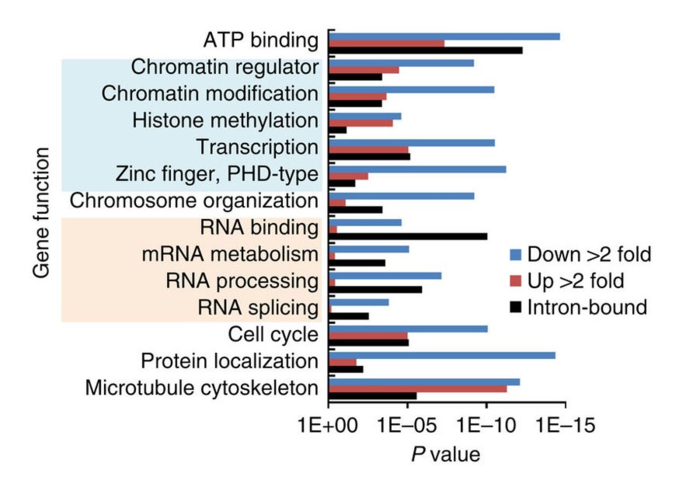
综上, AKAP95可能通过直接和间接调节染色质,转录和RNA加工调节全局基因表达。
拓展提高: 写一个Python脚本下载GEO数据
下载数据的过程无非是根据GEO找到FTP的地址,然后用wget或者prefetch下载而已。在我们今后的生涯里必然会遇到很多次类似的情况,所以写个脚本吧。
脚本逻辑很简单:
- 根据GEO accession找到FTP地址
- 用wget循环下载FTP地址下的数据
#!/bin/python3
import re
from urllib.request import urlopen
import os
def main(geo):
# find the FTP address from https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GEO
response = urlopen("https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc={}".format(geo))
pattern = re.compile("<a href=\"(.*?)\">\(ftp\)</a>")
# use wget from shell to download SRA data
ftp_address = re.search(pattern,response.read().decode('utf-8')).group(1)
os.system(' wget -nd -r 1 -A *.sra ' + ftp_address)
if __name__ == '__main__':
from sys import argv
main(argv[1])保存命名为SRR_downloader.py,在命令行里运行
python3 SRR_downloader.py GSE81916
简单说明:
- 用sys.argv从命令行中读取参数
- 用urllib.request向网页发起请求,获取response
- 用正则表达式(re)提取FTP地址
- 用os.system运行shell的命令
1F
转录组入门(2):读文章拿到测序数据