

# 进入你的source目录。
#*原文为cd ~/srrc,应是笔误,这里更正为:
cd ~/src
# 下载 SRA toolkit (确保你的下载链接对应的软件版本是跟你的系统一致的。)
#*建议安装最新版本:
#Mac
curl -O https://ftp-trace.ncbi.nlm.nih.gov/sra/sdk/2.8.2/sratoolkit.2.8.2-mac64.tar.gz
# Linux.
curl -O https://ftp-trace.ncbi.nlm.nih.gov/sra/sdk/2.8.2/sratoolkit.2.8.2-ubuntu64.tar.gz
#*以下命令,译者皆将旧版本改为新版本名称,如想继续安装旧版本,可以按着原英文版本安装。
#*原文版本是:
# Mac OSX.
curl -O http://ftp-trace.ncbi.nlm.nih.gov/sra/sdk/2.3.5-2/sratoolkit.2.3.5-2-mac64.tar.gz
# Linux.
curl -O http://ftp-trace.ncbi.nlm.nih.gov/sra/sdk/2.3.5-2/sratoolkit.2.3.5-2-ubuntu64.tar.gz
# 解压。(译者注:这是mac上的)
tar xzvf sratoolkit.2.8.2-mac64.tar.gz
#*方便起见,译者也给出Linux的:
tar xzvf sratoolkit.2.8.2-ubuntu64.tar.gz
# 转到这个目录下,并查看里面的文件。
cd sratoolkit.2.8.2-ubuntu64/
#*mac上
#*cd sratoolkit.2.8.2-mac64/
# 程序都在这个bin目录下
cd bin
# 来看看有什么。
ls
# 我们来把这些路径永久加到我们的PATH下。
#*前面提到export PATH=$PATH:~/src/edirect
#*这个命令只能是目前可以用,一旦退出系统,再重新进入,系统就不识别了。这里作者开始介绍如何使之变成每次进入这个终端都可以使用。
# 有一个特殊的文件在shell开启时会被系统自动读入。它为你开启的任一终端都提供好这些设置。这个文件在Mac上叫~/.profile,在linux上叫 ~/.bashrc 。 “ >> ”符号是指,把内容追加到一个文件后面,而非直接覆盖这个文件的原内容。你也可以通过text编辑器来编辑这个文件。
# Mac上:
echo export PATH=$PATH:~/src/edirect:~/src/sratoolkit.2.8.2-mac64/bin >> ~/.profile
# Linux上:
echo export PATH=$PATH:~/src/edirect:~/src/sratoolkit.2.8.2-ubuntu64/bin >> ~/.bashrc
# 你需要重新开启一个新的终端来使得上面的设置生效,或者你也可以:
source ~/.profile
#*译者温馨提醒:如果是Linux的话:source ~/.bashrc
# 转到 lecture 文件夹下。 -p 是用来做什么的?请看手册。
#* 可以用man mkdir查看mkdir的使用手册。
mkdir -p ~/edu/lec4
cd ~/edu/lec4
# 命令prefetch在哪里?
which prefetch
# 命令prefetch 可以从远程站点下载文件。
#来看一下帮助文档。
prefetch -h
# 来获取文件。
prefetch SRR1553610
# 这些文件去哪里了?存在了你home目录下的一个默认文件夹里。
ls ~/ncbi
# 里面添加了什么?可以用工具find来查看。
find ~/ncbi
# 我们用程序fastq-dump来把文件拆包
#*从NCBI下下来的数据,双端测序数据是放在一个文件里的,所以需要把它们重新拆解为两个文件。
fastq-dump -h
# 拆包文件
fastq-dump --split-files SRR1553610
# FASTQ格式的原始数据文件已经在当前文件夹了。
#*FASTQ格式在之前《小白生信学习记3》中有较为详细的介绍。但NCBI上下的fastq可能会跟测序得到的略为不同。
ls
# shell下的模式匹配。* (星号)表示可以匹配任何东西。
wc -l *.fastq
# 查看文件。
head SRR1553610_1.fastq
cat *.fastq | grep @SRR | wc -l
# 如何下载多个文件?创建一个含有SRR runs的文件。
echo SRR1553608 > sra.ids
echo SRR1553605 >> sra.ids
# 用这个文件去prefetch对应的runs.
prefetch --option-file sra.ids
# 拆包下载好的所有文件。请注意下边的做法不是特别妥当,因为(文件夹里)除了我们用sra.ids下载的,可能还有别的prefetch下来的文件。
fastq-dump --split-files ~/ncbi/public/sra/SRR15536*
# 正确的解法是,我们只拆包sra.ids里指定的文件。哎,但是fastq-dump不能直接实现。无语!
# 正确的解决这个问题,需要使用命令行作一些字符串处理
# 这会用到sed (字符流编辑器) 工具来提取文件并替换其中的模式“SRR”
cat sra.ids | sed 's/SRR/fastq-dump --split-files SRR/'
# 现在,把输出传到bash
cat sra.ids | sed 's/SRR/fastq-dump --split-files SRR/' | bash
# 我们还要做的更好。 为什么要去一一复制这些SRR ID ?我们其实可以完全通过命令行来获取它们。
# 然而efetch程序有一个bug。它获取到数据,但在结尾还会有一个error,为啥?很可能就是有个bug!无语! x 2
esearch -db sra -query PRJNA257197 | efetch -format runinfo
# 这个命令是把所有的结果放到一个文件里。
esearch -db sra -query PRJNA257197 | efetch -format runinfo > runinfo.txt
# 并且,由于这是一个逗号分隔符的文件,我们需要把分隔符(用以区别不同列的符号)指明给cut程序。
cat runinfo.txt | cut -f 1 -d ","
# 似乎所有的文件都在这了。这次搜索获得了195个文件(译者注:其实今天这个搜索结果已经到了891个了,下边我会直接使用891这个数据而不再提示)。
# 问: 我们相信这个结果吗?我们可以通过网站BioProject来获取文件,但你需要在正确的位置来实现这个功能。(文末译者会给出网站上如何获取这个list的方法。)
cat runinfo.txt | cut -f 1 -d ',' | grep SRR | wc -l
# 我们相信这个结果。但是可能你会有种不安,感觉自己可能犯错了。欢迎加入组织!
#*在我给出的网页版下载listd的方法的截图里,你也可以看到,最终结果确实是891个,跟命令行获取的是一样的。
cat runinfo.txt | cut -f 1 -d ',' | grep SRR > sra.ids
# 警告! 这会下载很多文件 (精确来说是891个).
# 目测这网速估计需要好几个小时。(译者注:这是作者基于195个结果作的判断)
prefetch --option-file sra.ids
#*如果你也想尝试一下下载,但又不想下载这么多。你可以这么做:
#*head sra.ids > sra_test.ids
#*prefetch --option-file sra_test.ids
# 检查一下下载结果的总大小
du -hs ~/ncbi
# 来把它们都转换了
cat sra_test.ids | sed 's/SRR/fastq-dump --split-files SRR/' | bash
# 这所有的文件一共有多少行?
wc -l *.fastq
# 通过这种方式,我们得到了PRJNA257197这个项目下所有的测序数据。
#*获取一个project里的所有SRR ID
#*首先进入https://www.ncbi.nlm.nih.gov/sra/
#*输入你要找的这个编号:PRJNA257197
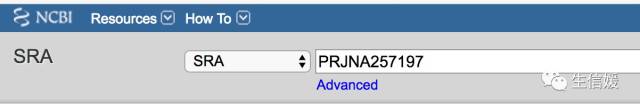
#*点击search
#*会看到很多检索结果。
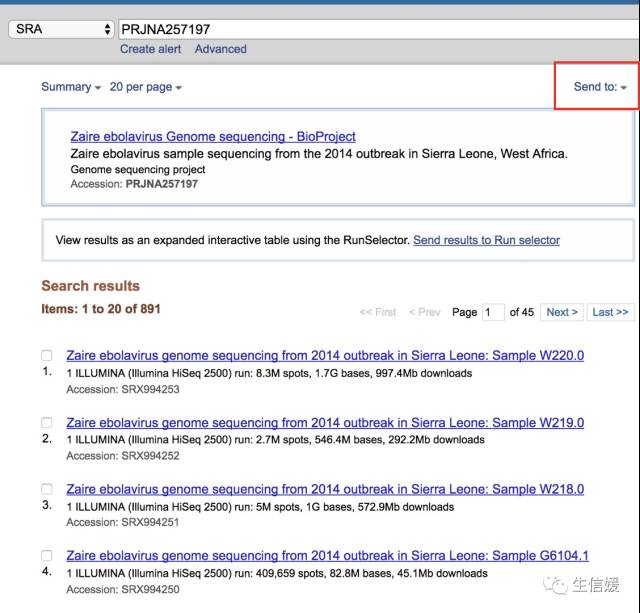
#*点击右上角的send to
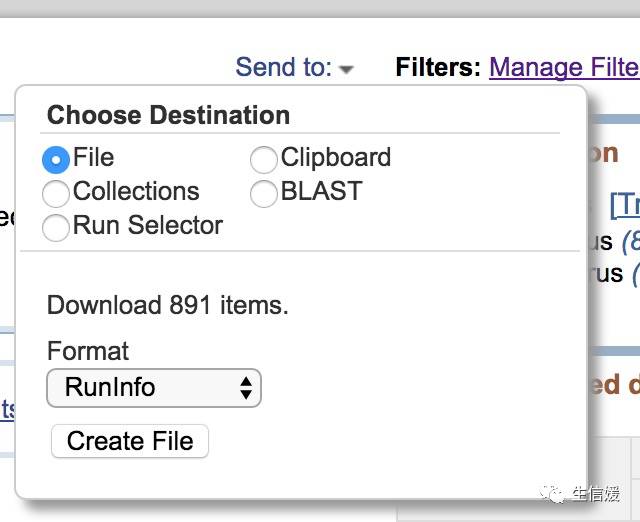
#*选定File,并把Format改为RunInfo
#*点击Create File就生成了一个SraRunInfo.csv文件了。
#*有没有发现,你其实只是把这种网页版的操作变成了几乎一一对应的命令行操作而已。