

写在前面
文献中常用的有DREME和HOMER,这次先搞定DREME,下次再写HOMER。
使用MEME套件中的DREME,用于鉴定meRIP-Seq数据中peak的motif。motif是序列中反复出现的由几个碱基组成的小片段,可能有特定的生物学意义。不过看meRIP-seq的文章里,这块的分析在结果中一般体现为一句话,交代找到的显著motif与已有报道一致,或者说多个发育阶段鉴定的保守motif是什么。
DREME的原理(http://meme-suite.org/doc/dreme-tutorial.html?man_type=web):
- 输入文件:positive sequences和negative sequences(背景序列,如果没有提供,会根据positive sequences自动生成);
- 寻找positive sequences中的3-8bp(默认长度)片段, 计算每个片段在positive和negative中出现的次数,通过Fisher's exact test 确定显著性,按照P-value排序成regular expression motifs.
- 选取前100个motif,将其中1个碱基替换成ambiguity code,重新计算p-value;重复该过程,使得每个碱基都可以有ambiguity code,得到generalized RE motifs.(相当于consensus sequences)
- 针对上一步得到的generalized RE motifs,计算每个motif在positive和negative中出现的次数,通过Fisher's exact test 确定显著性,按照设定的E-value值输出motif及相应的频率矩阵。
实战
01 安装
下载软件:http://meme-suite.org/doc/download.html
#install
mkdir meme
tar zxf meme_5.0.1.tar.gz
cd meme_5.0.1
./configure --prefix=/root/cc/biosoft/bin/meme --with-url=http://meme-suite.org --enable-build-libxml2 --enable-build-libxslt
make
make test
make install
#加入PATH(~/.bash_profile)
PATH=/root/cc/biosoft/bin/meme/bin:$PATH
source ~/.bash_profile
#test
dreme
#根据提示需要安装imagemagick
yum install ImageMagick
convert -version
02 实战
输入文件:fasta格式的序列文件
dreme -p input.fa -oc outDir -dna -png -eps -e 0.05 -t 18000 -m 20
#-dna, -rna : 默认是dna
#-png, -eps : 输出图片格式
# -e, -t, -m : 是设置软件结束运行的参数,不设置的话,默认在e-value>e时停止,否则会一直运行下去。
# -e: e-value,0.05(default) 一直search,直到下一个motif 的e-value >e
# -t: 运行时间 一直search,直到达到这个时间
# -m: 找到的motif数目 一直search,直到找到的motif数目达到这个值
#还可以设置Core Motif Width:--mink 最小值;--maxk 最大值
设置为-dna时,可以识别U(等同于T),但输出LOGO时使用T.
设置为-rna时,仍然能识别T(等同于U),但输出LOGO时使用U.
同样的序列和参数,设置为-dna和-rna,找到的motif是不一样的。Why? 没找到说明
是不是灰常简单?嘻嘻(#.#)
03 LOGO编辑
DREME生成的xml文件中,有每个motif的PWM matrix,里面有motif的每个位置上每种碱基的probability。可以用来直接画LOGO,方便的工具有如下几个。
seqLogo可以调整横纵坐标、碱基高度按比例/绝对大小展示等。
source("https://bioconductor.org/biocLite.R")
biocLite("seqLogo")
require(seqLogo)
mFile <- read.table("inp")
mFile
# V1 V2 V3 V4 V5 V6
#1 0 0.000000 1 0 1 0.000000
#2 0 0.000000 0 1 0 0.000000
#3 1 0.907213 0 0 0 0.636825
#4 0 0.092787 0 0 0 0.363175
p<-makePWM(m)
seqLogo(p)
seqLogo(p,ic.scale=FALSE)
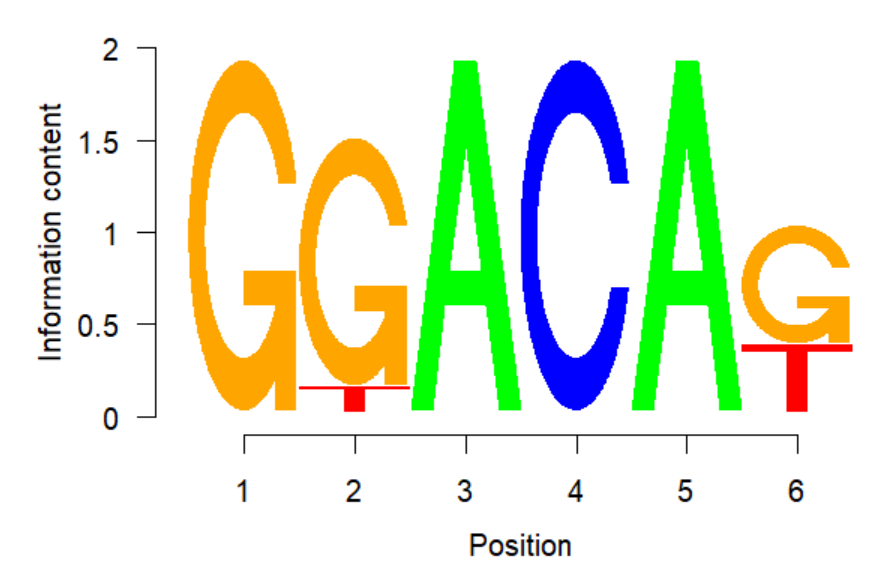
seqLogo(p)

seqLogo(p,ic.scale=FALSE)
不足:目前seqLogo只能识别DNA alphabet,无法识别RNA,Protein。
motifStack用途更广泛,这里仅用于把motif中的T替换成U。
需要安装ghostscript,然后将安装路径设置为环境变量R_GSCMD.
比如我安装的是64位的,安装在C:\Program Files\gs\gs9.23,按如下设置:
Sys.setenv(R_GSCMD=file.path("C:", "Program Files", "gs",
"gs9.23", "bin", "gswin64c.exe"))
将矩阵中的T换成U。
source("https://bioconductor.org/biocLite.R")
biocLite("motifStack")
require(motifStack)
Sys.setenv(R_GSCMD=file.path("C:", "Program Files", "gs",
"gs9.23", "bin", "gswin64c.exe"))
pcm=read.table("inp")
#0 0.000000 1 0 1 0.000000
#0 0.000000 0 1 0 0.000000
#1 0.907213 0 0 0 0.636825
#0 0.092787 0 0 0 0.363175
rownames(pcm) <- c("A","C","G","U")
motif <- new("pcm", mat=as.matrix(pcm), name="KD")
plot(motif,ncex=2,xaxis=F) #ncex: fond size of name; xaxis 不显示横坐标
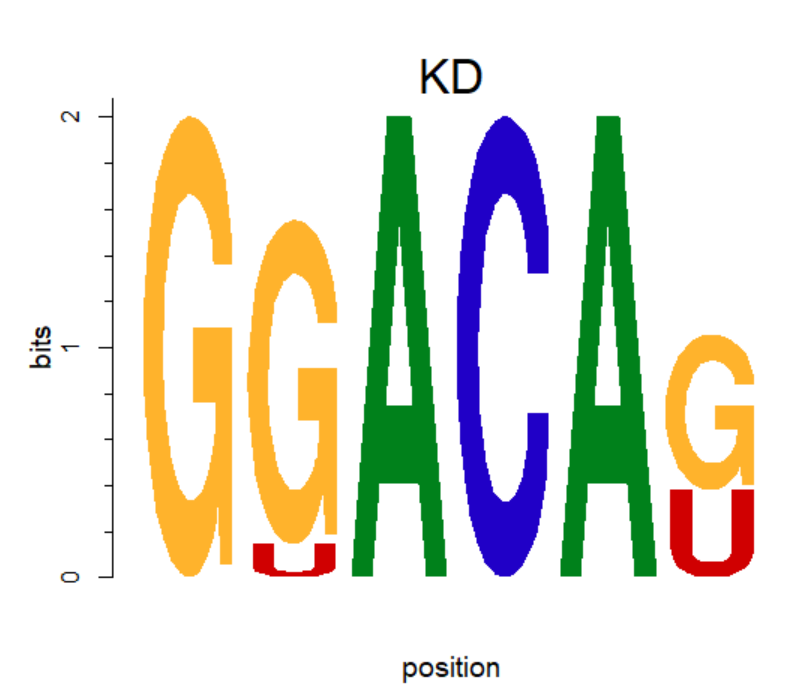
T--> U
04 Note
meRIP-seq的数据,分析得到的peak有时会跨区域。比如两个相邻的exon都是peak,但是得到的是一个包含这两个exon的区域,那么,提取序列时,应该提取的是真实的peak区域,即相应的exon区域。如果是exomePeak的结果,生成的BED12文件里包含block的信息,可以利用bedtools getfasta -split直接提取相应block的序列,再“串联”起来;如果是普通的BED文件(chr start end),那么就需要结合exon的坐标(比如GTF,GFF等注释文件),自己生成相应的BED12文件了。