

# 安装和使用Entrez Direct软件套件
# 到home目录下。
cd
# 等同于用~/ ,~/ 表示你的home目录。
cd ~/
# 创建src目录来存放这个课程里需要安装的程序。
#*你也可以创建为其他名字。如果自己修改了,那么下边的命令中记得对应修改。
mkdir ~/src
# 进入src目录
cd ~/src
# 获取entrez direct 工具包,大写 -O 是使得curl 命令去识别url上的文件名(作为下载后的文件名字)。
curl ftp://ftp.ncbi.nlm.nih.gov/entrez/entrezdirect/edirect.zip -O
# 上一条命令等同于:
curl ftp://ftp.ncbi.nlm.nih.gov/entrez/entrezdirect/edirect.zip -o edirect.zip
# 解压这个工具包.
unzip edirect.zip
# 查看新工具。
cd edirect
ls
# 为了使你的系统知道这些新工具在哪里,你需要把路径加到你的环境中,有多种方法可以实现。现在来把所有工具一次性加入到PATH变量里。
#*下面这个命令只能是目前可以用,一旦退出系统,再重新进入,系统就不识别了。如何使得每次开机都可以使用?下面会有讲。
export PATH=$PATH:~/src/edirect
#*可能是由于版本缘故,按照作者这个(2014年可行)的方法做,会出错。建议大家根据我的方法来做。
#*从这里开始是译者的方法
./setup.sh
#* 稍等一会后,会出现很多提示,其中有:Entrez Direct has been successfully downloaded and installed,以及类似这样的一行:
echo "export PATH=\$PATH:\$HOME/src/edirect" >> $HOME/.bashrc
#* 你可以直接复制这句话,然后粘贴到命令提示符后,回车运行。(如果是mac,建议把上句末尾的.bashrc改成.profile)
esearch -help
都会跳出对应的帮助文档。
这个软件具体的功能项可以查看https://www.ncbi.nlm.nih.gov/books/NBK179288/下的Entrez Direct Functions。

#*.bashrc 这个文件主要保存个人的一些个性化设置,如命令别名、路径等。
#*译者的安装方法,结束。
# 为这节课创建一个文件夹
mkdir -p ~/edu/lec3
cd ~/edu/lec3
pwd
# 运行 einfo
einfo -help
# 抓取描述信息,然后查看它们。
einfo -dbs > einfo-dbs.txt
more einfo-dbs.txt
einfo -db sra > einfo-sra.txt
more einfo-sra.txt
# 运行esearch。
esearch -help
esearch -db nucleotide -query PRJNA257197
esearch -db sra -query PRJNA257197
esearch -db protein -query PRJNA257197
#*译者注:-db是指定数据库类型,而query是跟着你要搜索的关键词。esearch -db nucleotide -query PRJNA257197这条命令的意思就是,在nucleotide这个数据库(database,简称db)里用关键词PRJNA257197搜索。以此类推。
#*不知什么原因,这个出来的结果不是很好看。
#*我来给大家解释一下这个运行结果。
#*以 esearch -db nucleotide -query PRJNA257197 这条命令为例
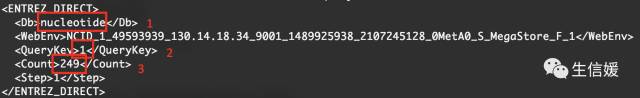
1:数据库类型为nucleotide,我们esearch的时候就指明了,我们要在nucleotide这个数据库=里搜索
2:搜索关键词数量:1(因为我们只输入了一个关键词,就是PRJNA257197)
3:搜索得到的条目有249个。
#*这么解释一下有没有感觉好点?
#*这一步相当于你到NCBI的首页,做了下图的操作:
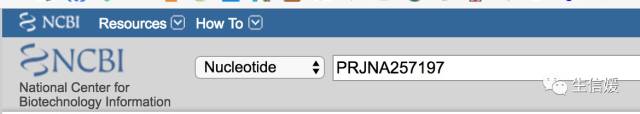
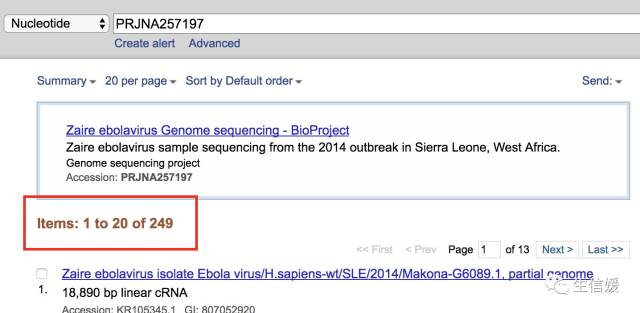
#*并点了Search
#*看!果然是249条检索结果。
# 抓取nucleotides数据。
#*译者注:前面只是做了搜索,但是要把数据弄下来,我们需要进一步用别的方式获取并存储下来。这里是用efetch把这249个搜索结果的fasta(也就是碱基序列)给存到了ebola.fasta里面。-format是指定格式,这里指定为fasta
esearch -db nucleotide -query PRJNA257197 | efetch -format fasta > ebola.fasta
# 来看一下这个文件里有多少条序列
#*fasta格式的一个特点就是以>开头,后面跟着序列的相关信息(没有也无妨)。下一行才是序列。fasta有时候也简称为fa。
cat ebola.fasta | grep ">" | wc -l
# 以GenBank格式获取数据
esearch -db nucleotide -query PRJNA257197 | efetch -format gb > ebola.gb