

1. CISA 简介
CISA (Contig Integrator for Sequence Assembly). 主要用于细菌基因组(小基因组)的整合。该软件使用简单,算法较好。
CISA 软件官网:http://sb.nhri.org.tw/CISA/en/CISA
2. CISA 算法
CISA分4步进行,如下图所示:
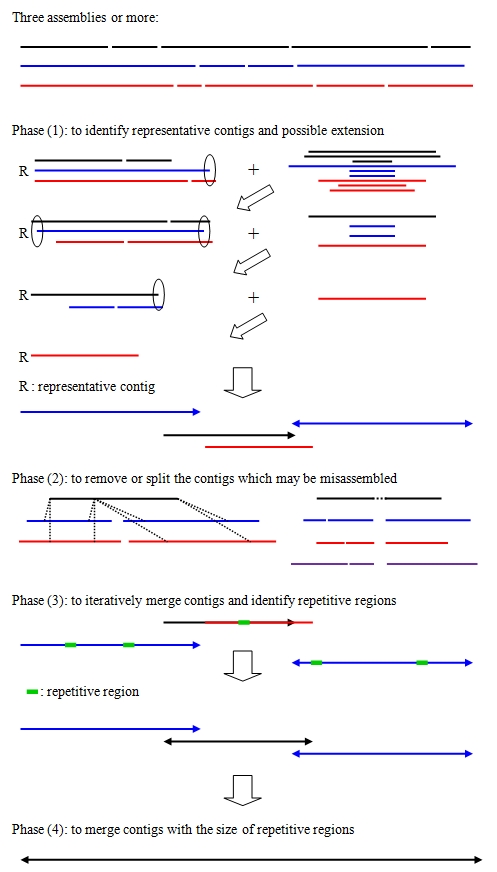
2.1 鉴定代表性的 contigs 并对其进行延伸
首先,将所有 Assemblies 的 contigs 进行合并,并按长度进行排序。然后调用 NUCmer 软件按从长到短的顺序对序列逐个进行基因组比对。通过此方法来得到序列在各个 Assemblies 之间的重叠状况,并将结果写入到 CISA1/explained.txt(序列重叠信息) 和 CISA1/Extend_info(序列延伸信息) 文件中。
设定的阈值为:非代表性序列与代表性序列的比对结果达到 >95% 覆盖度 和 >95% 的一致性; 或非代表性序列超出了代表性序列的两端时,比对结果 >80% 覆盖度 和 >95% 的一致性。
通过这种方法,最终得到延长的代表性序列,输出到 R1_Contigs.fa 文件中。
如图所示:对 3 个 Assemblies 进行 CISA 分析。首先找到所有 contigs 序列中最长的 contig 序列(蓝色线条表示)进行全基因组比对,得到了第 1 轮结果;然后使用剩下的序列(+号后面的序列)挑出最长的 contig 序列(蓝色线条表示),对剩下的序列使用 NUCmer 进行比对,得到第 2 轮结果; 继续使用剩下的序列挑出最长的 contig 序列(黑色线条表示),最为代表性序列进行比对;最终得到了 4 条(蓝蓝黑红)代表性序列,其中的空心椭圆和实心箭头分别代表延伸的位置和方向。
2.2 对 misassembled 的 contigs 进行去除或截断, 并对 contigs两端不确定区域进行截短
将 R1_Contigs.fa 的序列使用 NUCmer 比对到所有的 contigs 上。通过比对结果,从而对 misassembled 的 contigs 进行去除或截断, 或对 contigs两端不确定区域进行截短。
如图所示:
Phase(2)左图表示 1 条代表性序列比对到了 2 个不同的 contigs 的中间,则表明该代表性序列是错误的组装,则去除该代表性序列,同时引进相应的能比对到该代表性序列的 contigs。这些信息在 CISA2/Remove_Info 文件中。
Phase(2)右图表示 1 条代表性序列中间区域没有对应的 contigs 能匹配,则表明该序列是错误的组装,则将该区域进行去除,截断该代表性序列。有关Gaps 的信息在 CISA2/Gaps 文件中,若 Gap 长度大于其代表性序列长度的 95%,则去除该序列。
此外,对代表性序列首尾不确定性区域的信息在 CISA2/clip_info 和 CISA2/clip_out 文件中。
最终,对 misassembled 的 contigs 进行处理后,得到结果文件 R2_Contigs.fa。该文件的序列总长度偏大。
2.3 通过 Overlap 信息将 contigs 融合
如果 2 个 contigs 有 end-to-end 重叠,并且重叠区对较小 contigs 长度的比例 >30%, 则将其进行融合。使用多轮 blastn 进行迭代来融合 contigs 序列。同时也进行重复序列鉴定。
融合信息写入到每轮文件夹下的 Extend_info 文件中,同时每轮生成 temp.fa 文件,作为下一轮 blastn 的数据库构建的输入文件。
重复序列信息文件写入到每轮文件夹下的 Repeat_Region.txt 文件中。
2.4 通过 small overlap 进行 contigs 融合
上一步骤将 contigs 进行融合需要 >30% 覆盖度的 overlap,同时也鉴定了重复序列。如图所示:Phase3中,黑色与红色线条重叠区比例大,被整合到一起,该序列和其他两条蓝色序列的overlap比较small,不能进行融合。但是鉴定了其重复序列(绿色块表示)后,通过本步骤,若重复区域长度小于其 small overlap,则能将 contigs 进行融合。
此步骤和上一步骤类似,使用 blastn 进行多轮运算。同时将结果写入到每轮文件夹的 Extend_info 和 temp.fa 文件中。
最终的输出文件为 CISA.fa (由 cisa.config 配置文件指定的名称)
3. CISA 的下载与安装
$ wget http://sb.nhri.org.tw/CISA/upload/en/2014/3/CISA_20140304-05194132.tar $ tar xf CISA_20140304-05194132.tar -C /opt/biosoft
4. CISA 的使用
4.1 创建 merge.config 配置文件
配置文件如下:
count=3 data=abyss.fasta,title=abyss data=velvet.fasta,title=velvet data=allpathslg.fasta,title=allpathslg Master_file=merge_contigs.fa min_length=100 (default:100) Gap=11
4.2 Merge.py
$ /opt/biosoft/CISA1.3/Merge.py merge.config
根据上面 merge.config 配置文件信息,对 3 个 Assemblies 的 contigs 序列进行提取,并整合到 merge_contigs.fasta 文件中,同时输出各个 Assemblies 的 contigs size。
4.3 创建 cisa.config 配置文件
配置文件内容如下:
genome=47814562 (填写Assemblies中最大的 contigs size) infile=merge_contigs.fa outfile=cisa.contig.fa nucmer=/opt/biosoft/MUMmer3.23/nucmer R2_Gap=0.95 (default:0.95) CISA=/opt/biosoft/CISA1.3 makeblastdb=/opt/biosoft/ncbi-blast/bin/makeblastdb blastn=/opt/biosoft/ncbi-blast/bin/blastn
4.4 CISA.py
$ /opt/biosoft/CISA1.3/CISA.py cisa.config
启动主程序进行运算。基因组越大,Assemblies 数目越多,耗时越多(呈几何级数增加)。
5. 思考
CISA 应该要修改下,在第 2 步进行并行化运算,以提高速度。
可以分批进行两两融合,直到最终融合。多个两两融合进行并行化运算。
进行大基因组的融合时,CISA 不适用了。
原文来自:http://www.chenlianfu.com/?p=2135