

一.下载并安装这个软件
下载地址进下面,但是下载源码安装总是很困难,我直接下载bin文件可执行程序。
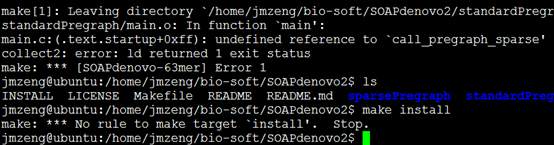
解压进入目录
首先make
然后make install即可
安装总是失败,我也不知道怎么回事,懒得解决了。
直接去我老师那里把这个程序拷贝进来了。
https://github.com/aquaskyline/SOAPdenovo2/archive/master.zip
http://sourceforge.net/projects/soapdenovo2/files/latest/download?source=files
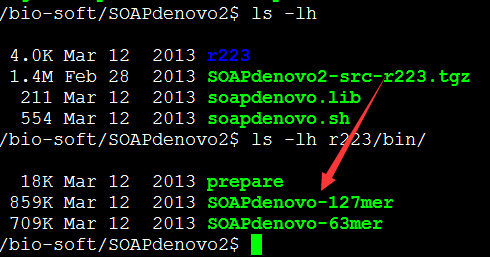
也可以直接下载bin程序
二.准备测试数据
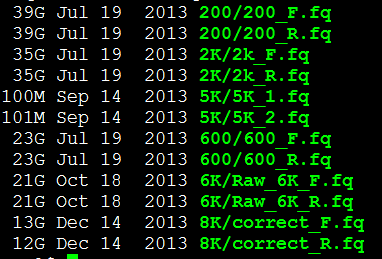
类似于这样的几个文库的左右两端测序数据。
我这里用一个小样本的单端数据做测试
三,参考命令
You may run it like this:
参考:https://www.plob.org/article/2537.html
https://github.com/aquaskyline/SOAPdenovo2
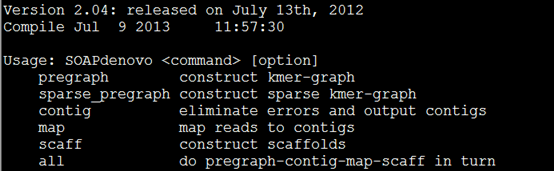
总共就四个步骤,介绍如下。
./pregraph_sparse [parameters]
./SOAPdenovo-63mer contig [parameters]
./SOAPdenovo-63mer map [parameters]
./SOAPdenovo-63mer scaff [parameters]
i) preparing the pregraph. This step is similar to velveth for velvet.
ii) Determining contigs. This step is similar to velvetg for velvet.
iii) Mapping back reads on to contigs.
iv) Assembling contigs into scaffolds.
SOAPdenovo-63mer sparse_pregraph -s config_file -K 45 -p 28 -z 1100000000 -o outPG SOAPdenovo-63mer contig -g outPG SOAPdenovo-63mer map -s config_file -g outPG -p 28 SOAPdenovo-63mer scaff -g outPG -p 28
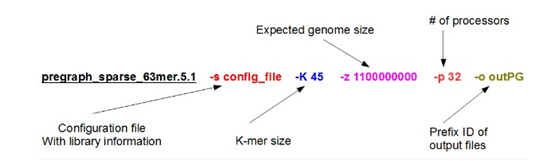
官网给出的步骤如下
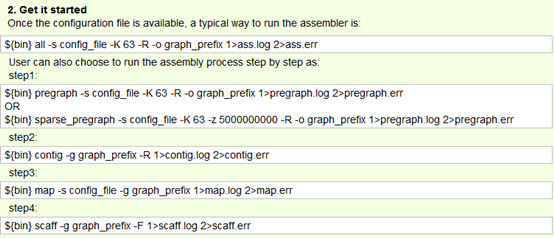
这个命令还需要一个配置文件
max_rd_len=99 设置最大reads长度,具体情况具体定义
[LIB] 第一个文库数据
avg_ins=225
reverse_seq=0
asm_flags=3
rank=1
q1=runPE_1.fq
q2=runPE_2.fq
[LIB] 第二个文库数据
avg_ins=2000
reverse_seq=1
asm_flags=2
rank=2
q1=runMP_1.fq
q2=runMP_2.fq
也可以全部一次性的搞一个命令
all -s config_file -K 63 -R -o graph_prefix 1>ass.log 2>ass.err
我简单修改了一下参考博客的代码跟官网的代码,然后运行了我自己的代码
/home/jmzeng/bio-soft/SOAPdenovo2-bin-LINUX-generic-r240/SOAPdenovo-127mer all -s config_file -K 63 -R -o graph_prefix 1>ass.log 2>ass.err
反正我也不懂,就先跑跑看咯
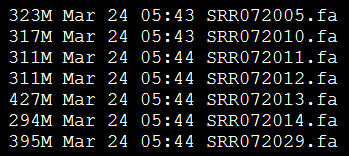
我选取的是7个单端数据,所以我的配置文件是
max_rd_len=500
[LIB]
avg_ins=225
reverse_seq=0
asm_flags=3
rank=1
p=SRR072005.fa
p=SRR072010.fa
p=SRR072011.fa
p=SRR072012.fa
p=SRR072013.fa
p=SRR072014.fa
p=SRR072029.fa
四.输出数据解读
好像我的数据都比较小,就7个三百多兆的fasta序列,几个小时就跑完啦
四个步骤都有输出数据
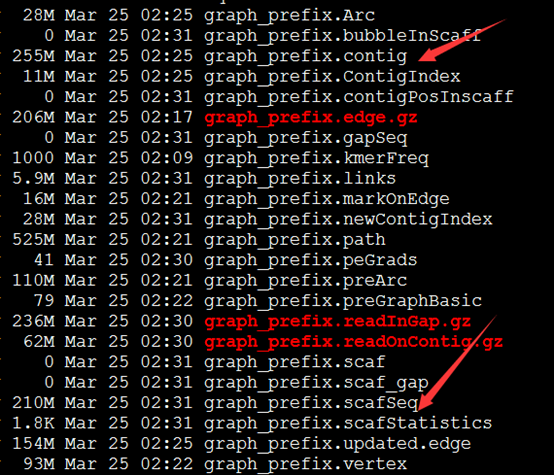
好像组装效果惨不忍睹呀!共86万的contig,50多万的scaffold
scaffolds>100 505473 99.60%
scaffolds>500 113523 22.37%
scaffolds>1K 48283 9.51%
scaffolds>10K 0 0.00%
scaffolds>100K 0 0.00%
scaffolds>1M 0 0.00%
这其实都相当于没有组装了,因为我的测序判断本来就很多是大于500的!
可能是我的kmer值选取的不对
Kmer为63跑出来的效果不怎么好,86万的contig,50万的scaffold的
Kmer为35跑出来的效果更惨,203万的contig,近60万的scaffold。
我觉得问题可能不是这里了,可能是没有用到那个20k和3k的双端测序库,唉,其实我习惯了illumina的测序数据,不太喜欢这个454的
感觉组装好难呀,业余时间搞不定呀,希望有高手能一起交流,哈哈,我自己再慢慢来试试。
原文来自:http://www.bio-info-trainee.com/476.html
1F
作者,你好,如你所说我下载的bin文件,有两个可执行程序,但是运行时,老提示命令不存在怎么回事?跪求解答 :wink: