

ANNOVAR简介
ANNOVAR是由王凯编写的一个注释软件,可以对SNP和indel进行注释,也可以进行变异的过滤筛选。
ANNOVAR能够利用最新的数据来分析各种基因组中的遗传变异。主要包含三种不同的注释方法,Gene-based Annotation(基于基因的注释)、Region-based Annotation(基于区域的注释)、Filter-based Annotation(基于筛选的注释)。
ANNOVAR由Perl编写。
优点:提供多个数据可直接下载、支持多种格式、注释直观;
缺点:没有数据库的物种无法注释。
ANNOVAR结构
ANNOVAR
│ annotate_variation.pl #主程序,功能包括下载数据库,三种不同的注释
│ coding_change.pl #可用来推断蛋白质序列
│ convert2annovar.pl #将多种格式转为.avinput的程序
│ retrieve_seq_from_fasta.pl #用于自行建立其他物种的转录本
│ table_annovar.pl #注释程序,可一次性完成三种类型的注释
│ variants_reduction.pl #可用来更灵活地定制过滤注释流程
│
├─example #存放示例文件
│
└─humandb #人类注释数据库
ANNOVAR下载数据库
命令示例
[kaiwang@biocluster ~/]$ Perl annotate_variation.pl -buildver hg19 -downdb -webfrom annovar refGene humandb/
# -buildver 表示version
# -downdb 下载数据库的指令
# -webfrom annovar 从annovar提供的镜像下载,不加此参数将寻找数据库本身的源
# humandb/ 存放于humandb/目录下
ANNOVAR的官方文档列出了可供下载的数据库及版本、更新日期等信息,可用-downdb avdblist参数查看。

ANNOVAR输入格式
[kaiwang@biocluster ~/]$ cat example/ex1.avinput 1 948921 948921 T C comments: rs15842, a SNP in 5\\' UTR of ISG15 1 1404001 1404001 G T comments: rs149123833, a SNP in 3\\' UTR of ATAD3C 1 5935162 5935162 A T comments: rs1287637, a splice site variant in NPHP4 1 162736463 162736463 C T comments: rs1000050, a SNP in Illumina SNP arrays 1 84875173 84875173 C T comments: rs6576700 or SNP_A-1780419, a SNP in Affymetrix SNP arrays 1 13211293 13211294 TC - comments: rs59770105, a 2-bp deletion 1 11403596 11403596 - AT comments: rs35561142, a 2-bp insertion 1 105492231 105492231 A ATAAA comments: rs10552169, a block substitution 1 67705958 67705958 G A comments: rs11209026 (R381Q), a SNP in IL23R associated with Crohn\\'s disease 2 234183368 234183368 A G comments: rs2241880 (T300A), a SNP in the ATG16L1 associated with Crohn\\'s disease 16 50745926 50745926 C T comments: rs2066844 (R702W), a non-synonymous SNP in NOD2 16 50756540 50756540 G C comments: rs2066845 (G908R), a non-synonymous SNP in NOD2 16 50763778 50763778 - C comments: rs2066847 (c.3016_3017insC), a frameshift SNP in NOD2 13 20763686 20763686 G - comments: rs1801002 (del35G), a frameshift mutation in GJB2, associated with hearing loss 13 20797176 21105944 0 - comments: a 342kb deletion encompassing GJB6, associated with hearing loss
ANNOVAR使用.avinput格式,如以上代码所示,该格式每列以tab分割,最重要的地方为前5列,分别是:
1. 染色体(Chromosome)
2. 起始位置(Start)
3. 结束位置(End)
4. 参考等位基因(Reference Allele)
5. 替代等位基因(Alternative Allele)
6. 剩下为注释部分(可选)。
ANNOVAR主要也是依靠这5处信息对数据库进行比对,进而注释变异。
ANNOVAR格式转换
命令示例
$ convert2annovar.pl -format vcf4 example/ex2.vcf > ex2.avinput # -format vcf4 指定格式为vcf
ANNOVAR主要使用convert2annovar.pl程序进行转换,转换后文件是精简过的,主要包含前面提到的5列内容,如果要将原格式的文件的所有内容都包含在转换后的.avinput文件中,可以使用-includeinfo参数;如果需要分开每个sample输出单一的.avinput文件,可以使用-allsample参数,等等。
ANNOVAR还主要支持以下格式转换:
- SAMtools pileup format
- Complete Genomics format
- GFF3-SOLiD calling format
- SOAPsnp calling format
- MAQ calling format
- CASAVA calling format
ANNOVAR注释功能
用table_annovar.pl进行注释(可一次性完成三种类型的注释)
命令示例
[kaiwang@biocluster ~/]$ table_annovar.pl example/ex1.avinput humandb/ -buildver hg19 -out myanno -remove -protocol refGene,cytoBand,genomicSuperDups,esp6500siv2_all,1000g2014oct_all,1000g2014oct_afr,1000g2014oct_eas,1000g2014oct_eur,snp138,ljb26_all -operation g,r,r,f,f,f,f,f,f,f -nastring . -csvout # -buildver hg19 表示使用hg19版本 # -out myanno 表示输出文件的前缀为myanno # -remove 表示删除注释过程中的临时文件 # -protocol 表示注释使用的数据库,用逗号隔开,且要注意顺序 # -operation 表示对应顺序的数据库的类型(g代表gene-based、r代表region-based、f代表filter-based),用逗号隔开,注意顺序 # -nastring . 表示用点号替代缺省的值 # -csvout 表示最后输出.csv文件
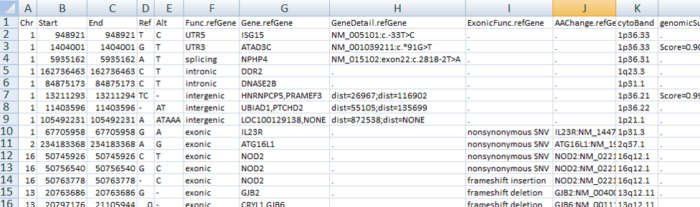
输出的csv文件将包含输入的5列主要信息以及各个数据库里的注释,此外,table_annoval.pl可以直接对vcf文件进行注释(不需要转换格式),注释的内容将会放在vcf文件的“INFO”那一栏。
Gene-based Annotation(基于基因的注释)
基于基因的注释(gene-based annotation)揭示variant与已知基因直接的关系以及对其产生的功能性影响,需要使用for gene-based的数据库。
命令示例
[kaiwang@biocluster ~/]$ annotate_variation.pl -geneanno -dbtype refGene -out ex1 -build hg19 example/ex1.avinput humandb/ # -geneanno 表示使用基于基因的注释 # -dbtype refGene 表示使用"refGene"数据库 # -out ex1 表示输出文件以ex1为前缀
因为annotate_variation.pl默认使用gene-based注释类型以及refGene数据库,所以上面的命令可以缺省-geneanno -dbtype refGene。
运行命令后将会生成3个文件:
- ex1.variant_function 注释所有变异所在基因及位置
- ex1.exonicvariantfunction 详细注释外显子区域的变异功能、类型、氨基酸改变等
- ex1.ann.log log文件,包含运行的命令行及运行提示,所用数据库文件
ex1.variant_function
第一个文件以.variant_function结尾,主要的内容如下
[kaiwang@biocluster ~/]$ cat ex1.variant_function UTR5 ISG15(NM_005101:c.-33T>C) 1 948921 948921 T C comments: rs15842, a SNP in 5\\' UTR of ISG15 UTR3 ATAD3C(NM_001039211:c.*91G>T) 1 1404001 1404001 G T comments: rs149123833, a SNP in 3\\' UTR of ATAD3C splicing NPHP4(NM_001291593:exon19:c.1279-2T>A,NM_001291594:exon18:c.1282-2T>A,NM_015102:exon22:c.2818-2T>A) 1 5935162 5935162 A T comments: rs1287637, a splice site variant in NPHP4 intronic DDR2 1 162736463 162736463 C T comments: rs1000050, a SNP in Illumina SNP arrays intronic DNASE2B 1 84875173 84875173 C T comments: rs6576700 or SNP_A-1780419, a SNP in Affymetrix SNP arrays intergenic LOC645354(dist=11566),LOC391003(dist=116902) 1 13211293 13211294 TC - comments: rs59770105, a 2-bp deletion intergenic UBIAD1(dist=55105),PTCHD2(dist=135699) 1 11403596 11403596 - AT comments: rs35561142, a 2-bp insertion intergenic LOC100129138(dist=872538),NONE(dist=NONE) 1 105492231 105492231 A ATAAA comments: rs10552169, a block substitution exonic IL23R 1 67705958 67705958 G A comments: rs11209026 (R381Q), a SNP in IL23R associated with Crohn\\'s disease exonic ATG16L1 2 234183368 234183368 A G comments: rs2241880 (T300A), a SNP in the ATG16L1 associated with Crohn\\'s disease exonic NOD2 16 50745926 50745926 C T comments: rs2066844 (R702W), a non-synonymous SNP in NOD2 exonic NOD2 16 50756540 50756540 G C comments: rs2066845 (G908R), a non-synonymous SNP in NOD2 exonic NOD2 16 50763778 50763778 - C comments: rs2066847 (c.3016_3017insC), a frameshift SNP in NOD2 exonic GJB2 13 20763686 20763686 G - comments: rs1801002 (del35G), a frameshift mutation in GJB2, associated with hearing loss exonic CRYL1,GJB6 13 20797176 21105944 0 - comments: a 342kb deletion encompassing GJB6, associated with hearing loss
注释后输出的文件,同样每列以tab分割,第1列为变异所在的类型,如外显子(exonic)、UTR5、UTR3等(官方文档有详细的类型列表)。
如果第1列的为外显子、内含子或者非编码RNA,第二行将是对应的基因名(有多个基因名则会以逗号隔开);否则第二列将会给出相邻的两个基因以及对应的距离。
从第3列开始至第7列为输入的那5列主要信息,剩余为注释信息。
需要注意的是,如果该变异找到多种注释,ANNOVAR将会对它进行比较,以exonic = splicing > ncRNA > UTR5/UTR3 > intron > upstream/downstream > intergenic 的优先权重,取最优的表示,如果你想ANNOVAR列出该变异所有注释,可以使用--separate参数。
ex1.exonic_variant_function
第二个输出文件以.exonic_variant_function结尾,只列出外显子(氨基酸会改变)的变异,主要内容如下
[kaiwang@biocluster ~/]$ cat ex1.exonic_variant_function line9 nonsynonymous SNV IL23R:NM_144701:exon9:c.G1142A:p.R381Q, 1 67705958 67705958 G A comments: rs11209026 (R381Q), a SNP in IL23R associated with Crohn\\'s disease line10 nonsynonymous SNV ATG16L1:NM_001190267:exon9:c.A550G:p.T184A,ATG16L1:NM_017974:exon8:c.A841G:p.T281A,ATG16L1:NM_001190266:exon9:c.A646G:p.T216A,ATG16L1:NM_030803:exon9:c.A898G:p.T300A,ATG16L1:NM_198890:exon5:c.A409G:p.T137A, 2 234183368 234183368 A G comments: rs2241880 (T300A), a SNP in the ATG16L1 associated with Crohn\\'s disease line11 nonsynonymous SNV NOD2:NM_022162:exon4:c.C2104T:p.R702W,NOD2:NM_001293557:exon3:c.C2023T:p.R675W, 16 50745926 50745926 C comments: rs2066844 (R702W), a non-synonymous SNP in NOD2 line12 nonsynonymous SNV NOD2:NM_022162:exon8:c.G2722C:p.G908R,NOD2:NM_001293557:exon7:c.G2641C:p.G881R, 16 50756540 50756540 G comments: rs2066845 (G908R), a non-synonymous SNP in NOD2 line13 frameshift insertion NOD2:NM_022162:exon11:c.3017dupC:p.A1006fs,NOD2:NM_001293557:exon10:c.2936dupC:p.A979fs, 16 50763778 5076377comments: rs2066847 (c.3016_3017insC), a frameshift SNP in NOD2 line14 frameshift deletion GJB2:NM_004004:exon2:c.35delG:p.G12fs, 13 20763686 20763686 G - comments: rs1801002 (del35G), a frameshift mutation in GJB2, associated with hearing loss line15 frameshift deletion GJB6:NM_001110221:wholegene,GJB6:NM_001110220:wholegene,GJB6:NM_001110219:wholegene,CRYL1:NM_015974:wholegene,GJB6:NM_006783:wholegene, 13 20797176 21105944 0 - comments: a 342kb deletion encompassing GJB6, associated with hearing loss
该文件的第1列为.variant_function文件中该变异所在的行号;第2列为该变异的功能性后果,如非同义SNV、同义SNV、移码插入等(官方文档同样有详细的类型列表);第3列包括基因名称、转录识别标志和相应的转录本的序列变化。第四列开始为输入文件的内容。
Region-based Annotation(基于区域的注释)
基于过滤的注释精确匹配查询变异与数据库中的记录:如果它们有相同的染色体,起始位置,结束位置,REF的等位基因和ALT的等位基因,才能认为匹配。基于区域的注释看起来更像一个区域的查询(这个区域也可以是一个单一的位点),在一个数据库中,它不在乎位置的精确匹配,它不在乎核苷酸的识别。
基于区域的注释(region-based annotation)揭示variant与不同基因组特定段的关系,例如:它是否落在已知的保守基因组区域。基于区域的注释的数据库一般由UCSC提供。
命令示例
[kaiwang@biocluster ~/]$ annotate_variation.pl -regionanno -build hg19 -out ex1 -dbtype phastConsElements46way example/ex1.avinput humandb/ # -regionanno 表示使用基于区域的注释 # -dbtype phastConsElements46way 表示使用"phastConsElements46way"数据库,注意需要使用Region-based的数据库
输出文件是ex1.hg19_phastConsElements46way,可以看到,Region-based 注释将会生成以注释数据库为后缀的注释文件。该文件主要内容有
[kaiwang@biocluster ~/]$ cat ex1.hg19_phastConsElements46way phastConsElements46way Score=387;Name=lod=50 1 67705958 67705958 G A comments: rs11209026 (R381Q), a SNP in IL23R associated with Crohn\\'s disease phastConsElements46way Score=420;Name=lod=68 16 50756540 50756540 G C comments: rs2066845 (G908R), a non-synonymous SNP in NOD2 phastConsElements46way Score=385;Name=lod=49 16 50763778 50763778 - C comments: rs2066847 (c.3016_3017insC), a frameshift SNP in NOD2 phastConsElements46way Score=395;Name=lod=54 13 20763686 20763686 G - comments: rs1801002 (del35G), a frameshift mutation in GJB2, associated with hearing loss phastConsElements46way Score=545;Name=lod=218 13 20797176 21105944 0 - comments: a 342kb deletion encompassing GJB6, associated with hearing loss
输出的注释文件第1列为“phastConsElements46way”,对应注释的类型,这里的phastCons 46-way alignments属于保守的基因组区域的注释;第二列包含评分和名称,评分来自UCSC,可以使用--score_threshold和--normscore_threshold来过滤评分低的变异,“Name=lod=x”名称表示该区域的名称;剩余的部分为输入文件的内容。
Filter-based Annotation(基于过滤的注释)
filter-based和region-based主要的区别是,filter-based针对mutation(核苷酸的变化)而region-based针对染色体上的位置。例如region-based比对chr1:1000-1000而filter-based比对chr1:1000-1000上的A->G。
基于过滤的注释,使用不同的过滤数据库,可以给出这个variant的一系列信息。如在全基因组数据中的变异频率,可使用1000g2015aug、kaviar_20150923等数据库;在全外显组数据中的变异频率,可使用exac03、esp6500siv2等;在孤立的或者低代表人群中的变异频率,可使用ajews等数据库。(在ANNOVAR官方文档中也有详细的介绍)
命令示例
[kaiwang@biocluster ~/]$ annotate_variation.pl -filter -dbtype 1000g2012apr_eur -buildver hg19 -out ex1 example/ex1.avinput humandb/ # -filter 使用基于过滤的注释 # -dbtype 1000g2012apr_eur 使用"1000g2012apr_eur"数据库
运行命令后,已知的变异会被写入一个*dropped结尾的文件,而没有在数据库中找到的变异将会被写入*filtered结尾的文件,*dropped文件是我们所需要的结果。这个文件内容如下
[kaiwang@biocluster ~/]$ cat ex1.hg19_EUR.sites.2012_04_dropped 1000g2012apr_eur 0.04 1 1404001 1404001 G T comments: rs149123833, a SNP in 3\\' UTR of ATAD3C 1000g2012apr_eur 0.87 1 162736463 162736463 C T comments: rs1000050, a SNP in Illumina SNP arrays 1000g2012apr_eur 0.81 1 5935162 5935162 A T comments: rs1287637, a splice site variant in NPHP4 1000g2012apr_eur 0.06 1 67705958 67705958 G A comments: rs11209026 (R381Q), a SNP in IL23R associated with Crohn\\'s disease 1000g2012apr_eur 0.54 1 84875173 84875173 C T comments: rs6576700 or SNP_A-1780419, a SNP in Affymetrix SNP arrays 1000g2012apr_eur 0.96 1 948921 948921 T C comments: rs15842, a SNP in 5\\' UTR of ISG15 1000g2012apr_eur 0.05 16 50745926 50745926 C T comments: rs2066844 (R702W), a non-synonymous SNP in NOD2 1000g2012apr_eur 0.01 16 50756540 50756540 G C comments: rs2066845 (G908R), a non-synonymous SNP in NOD2 1000g2012apr_eur 0.01 16 50763778 50763778 - C comments: rs2066847 (c.3016_3017insC), a frameshift SNP in NOD2 1000g2012apr_eur 0.53 2 234183368 234183368 A G comments: rs2241880 (T300A), a SNP in the ATG16L1 associated with Crohn\\'s disease
*dropped文件第1列如region-based注释的结果一样以数据库命名;第二列为等位基因频率,我们可以用-maf 0.05参数来过滤掉低于0.05的变异,;第三列开始同样是输入文件的内容。
需要注意的是,我们也可以使用-maf 0.05 -reverse过滤掉高于0.05的变异;但是过滤ALT等位基因的频率,我们更提倡使用-score_threshold参数。
ANNOVAR其他程序
ANNOVAR包里还有
- Variants_Reduction: prioritizing causal variants
- Coding_Change: Infer mutated protein sequence
- RetrieveSeqfrom_FASTA: Retrieve nucleotide/protein sequences
三个程序没有介绍,可以参考官方文档的Accessory Programs自行了解。
参考文献:
- Wang K, Li M, Hakonarson H. ANNOVAR: Functional annotation of genetic variants from next-generation sequencing data Nucleic Acids Research, 38:e164, 2010
- ANNOVAR Documentation
- annovar对人类基因组和非人类基因组variants注释流程
原文来自:http://zhengzexin.com/2016/04/28/annovar-zhu-shi-ruan-jian/