

什么是基因组变异
基因组变异是一个定义比较模糊的概念. 所谓的变异是相对于一个完美的“参考基因组”而言。但是其实完美的“参考基因组”并不存在,因为我们只是选择某一个物种里的其中似乎比较正常的个体进行测序组装,然后基于它进行后续的研究。简单的说,参考和变异是相对而言,变异也可能完全正常。
常见的基因组变异一般可以归为如下几类:
- SNP, 单核苷酸多态性, 一个碱基的变化
- INDEL,插入或缺失, 一个碱基的增加或移除
- SNV, 单核苷酸变异,一个碱基的改变,可以是SNP,也可以是INDEL
- MNP, 多核苷酸多态性,一个区块中有多个保守的SNP
- MNV,多核苷酸变异,一个区块中有多个SNP或INDEL
- short variations, 小于50bp的变异
- large-scale variation, 大于50bp的大规模变异
- SV, 结构变异,通常是上千个碱基,甚至是染色体级别上的变异
研究这些变异需要用到不同的手段,其中普通的DNA二代测序在寻找20bp以下的变异比较靠谱,对于大于1kb的结构变异而言,采用光学图谱(Bionanogenomics)可能更加靠谱一点。因此,对于目前最常用的二代测序而言,还是尽量就找SNP和INDEL吧,几个碱基的变化找起来还是相对容易些和靠谱些。
对于SNP的定义这里也要注意下,最初的SNP的定义指的是单个碱基导致的多态性,在群体中广泛存在(1%),可用来作为分子标记来区分不同个体。目前的定义比较粗暴一点,就是那个和“参考基因组”不同的单个位点。值得注意的是,这个概念可能不同人还有不同的定义,当你和别人就某个问题争执的时候,最好问问他是如何定义这个基本概念。由于SNP的广泛存在,并且变异可能会导致疾病,也就是存在某些SNP会导致疾病。Online Mendelian Inheritance in Man,就是一个人类遗传疾病数据库,建议去看下。
最后说下genotype和haplotype。genotype,基因型指的是一个个体的遗传组成。但是对于基因组变异而言,基因型通常指的是个体在某个位点上的等位基因情况。haplotype, 单倍型最初指的是从单个亲本中遗传的一组基因,而在基因组变异背景下,则是指一组变异。
一次简单的变异检测实战
变异检测(variant calling)即通过比较参考序列和比对结果来找到两者的不同并记录,基本上可以分为如下几步:
- 序列比对
- 比对后处理(可选)
- 从联配中确定变异
- 根据某些标准进行过滤
- 对过滤的变异注释
这里面的每一个可选的工具都有很多,不同工具组合后的分析流程得到的结果可能会有很大差异。在变异检测这一部分目前就有很多软件,但是常用并且相对比较可靠的工具有如下几个:
- bcftools: http://www.htslib.org/doc/bcftools.html
- FreeBayes: https://github.com/ekg/freebayes
- GATK: https://software.broadinstitute.org/gatk/
- VarScan2: http://varscan.sourceforge.net/
当然这些工具最初都是用于人类基因组。
以埃博拉基因组为例完成一次简单的Variant Calling,所需工具为efetch, fastq-dump, emboss/seqret,bwa, samtools和FreeBayes和snpEff。这些都可以通过conda快速安装。
第一步: 获取参考基因组序列,并建立索引
# 建立文件夹
mkdir -p refs
# 根据Accession下载
ACC=AF086833
REF=refs/$ACC.fa
efetch -db=nuccore -format=fasta -id=$ACC | seqret -filter -sid $ACC > $REF
bwa index $REF
第二步: 获取需要比对的测序数据, 以前10w条为例
# 仅要前10w条read
SRR=SRR1553500
fastq-dump -X 100000 --split-files $SRR
第三步:序列比对
BAM=$SRR.bam
R1=${SRR}_1.fastq
R2=${SRR}_2.fastq
TAG="@RG\tID:$SRR\tSM:$SRR\tLB:$SRR"
bwa mem -R $TAG $REF $R1 $R2 | samtools sort > $BAM
samtools index $BAM
第四步:使用freebayes或HaplotypeCaller(GATK4)检测变异
freebayes -f $REF $BAM > ${SRR}_freebayes.vcf
gatk HaplotypeCaller -R $REF-I $BAM -stand-call-conf 30 \
-bamout bamout.bam
--genotyping-mode DISCOVERY
-O ${SRR}_haplotypecaller.vcf
用IGV可视化的效果如下:
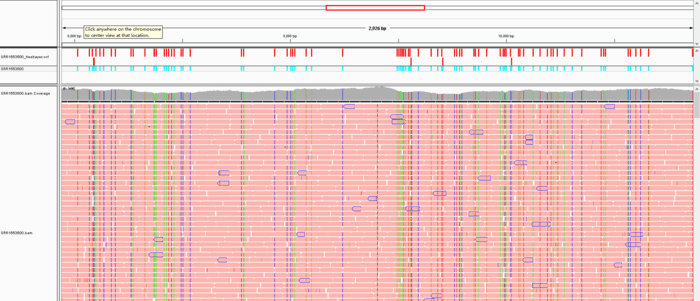
这是最简单的变异检测流程,对于找到的变异还可以进一步过滤,这一部分内容见call variant中关于snp筛选的一些思考
第五步:变异标准化(可选)
由于VCF文件的灵活性,同一种变异可以通过不同的形式表示, 如下图
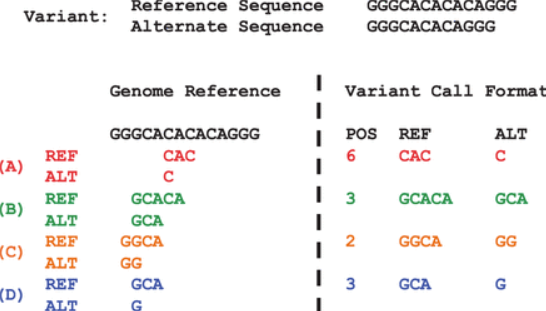
变异标准化按照如下规则对变异位点表示进行简化
- 尽可能以少的字符表示变异
- 无等位基因可以标识为长度为0
- 变异位点必须左对齐
看起来很复杂,其实操作起来很简单
bcftools norm -f $REF SRR1553500_freebayes.vcf > SRR1553500_freebayes_norm.vcf
# Lines total/split/realigned/skipped: 493/0/0/0
大部分软件,如GATK, freebayes已经是标准化的结果。
变异检测那么简单吗?
经过简单的实战之后,似乎变异检测是一件非常容易的事情,只要敲几行命令就行了。当然最开始我也是想的,毕竟无知者无畏,但是了解的越多,你就会发现事情并没有那么简单。**大部分基因组相关的DNA序列有一些特性是人类的直觉所不能理解的,因为这需要考虑一些背景。
- DNA序列可以非常的长
- A/T/G/C能够构建任意组合的DNA序列,因此在完全随机情况下,即使随机分配也能产生各种各样的模式。
- DNA序列只有部分会受到随机影响,基本上这部分序列都是有功能的。
- 不同物种的不同的DNA序列受到不同的随机性影响
- 我们按照实验流程将大片段DNA破碎成小的部分,并尝试通过和参考基因组比对找到原来的位置。只有它依旧和原来的位置非常靠近,才能进一步寻找变异。
因此即便序列和基因组某个序列非常接近,从算法的角度是正确比对,但其实偏离了原来正确的位置,那么从这部分找到的变异也是错误的。那我们有办法解决这个问题嘛?基本上不可能,除非技术进步后,我们可以一次性通读所有序列。当然目前比较常用的方法是找到最优变异,并且那个变异能更好的解释问题,且和每条read中的变异都是一致的。这就是目前变异检测软件常用策略:realignment或probabilistic alignment。
变异注释
变异注释意味着猜测遗传变异(SNP, INDEL, CNY, SV)对基因功能,转录本和蛋白序列以及调控序列的影响。为了对变异进行预测,预测软件需要你提供基因组注释信息,并且注释信息的完善程度决定了预测的准确性。变异预测一般会提供如下结果
- 变异位点所在基因组注释的位置,是转录本上游,还是编码区,还是非编码RNA
- 列举出收到影响的转录本和基因
- 确定变异在蛋白序列上的影响, 如stop_gained(终止密码子提前), missense(错义), stop_lost(终止密码子缺失), frameshift(移码)等
- 对于人类,还可以和已知的位点进行匹配
这些效应的定义可以在序列本体论查询.
变异注释常用软件有:VEP, snpEFF, AnnoVar, VAAST2. 其中VEP是网页工具http://asia.ensembl.org/Tools/VEP, 使用很方便,可惜支持的物种有限。snpEFF可以说是支持物种最多的工具,这里使用它。
snpEff databases > listing.txt
# 确认物种名
grep -i ebola databases
# 下载
snpEff download ebola_zaire
下载之后还需要检查一下snpEFF提供的注释是否和我们所使用参考基因组一致.
snpEff dump ebola_zaire | less
很不幸的是,snpEFF提供的是基于KJ660346构建的数据库,而我们使用的是AF086833。因此需要重新下载对应的参考基因组重新比对,进行注释。对饮参考基因组的地址为https://www.ncbi.nlm.nih.gov/nuccore/KJ660346.1?report=fasta
snpEff ebola_zaire ${SRR}_freebayes.vcf > ${SRR}_annotated.vcf
最终会生成注释后的VCF文件以及变异位点的描述性报告。
作者:
链接:
來源:简书
著作权归作者所有。商业转载请联系作者获得授权,非商业转载请注明出处。