

为什么需要本体论
作为一位大学统计棉花表皮毛的苦逼生物狗,深刻体会什么叫做经验,也就是人类模式识别能力的强大和不精确性。当时的导师教我如何根据表皮毛的长短和浓密进行基因型的判定,但是我一直纠结长和短,密和疏之间的分界。在读研的时候,师姐会让我提供基因Genomic序列,这来自于TAIR的定义,此外TAIR还定义了full length cDNA和full length CDS。我经常纠结这些序列和我GFF里面的CDS,mRNA,gene的关系是什么?直到我把所有序列都拿出来,进行多序列联配才发现它们之间的差异。
计算机科学来自于多学科的交互,比如说数学,语言学,逻辑学等。为了保证互联网的通信,代码的复用,API的调用等,计算机协会制定了很多协议进行标准化。比如说“意思意思”这句话在中文的语境千变万化,但是在计算机里面可能就会翻译成mean of mean。为了能让计算机分析生物数据,就要生物学的一些概念进行精确定义,而不是“只可意会,不可言传”
Karen在文章The Sequence Ontology: a tool for the unification of genome annotations就写了这样一段话:
Unfortunately, biological terminology is notoriously ambiguous; the same word is often used to describe more than one thing and there are many dialects. For example, does a coding sequence (CDS) contain the stop codon or is the stop codon part of the 3'-untranslated region (3' UTR)?
There really is no right or wrong answer to such questions, but consistency is crucial when attempting to compare annotations from different sources, or even when comparing annotations performed by the same group over an extended period of time.****
也就是说一致性比正确性更加重要,如果双方各执一见,对一个概念的定义模棱两可,那么讨论只会浪费时间。这就是为什么我们需要建立统一的概念。
本体论是什么
本体论是概念化的详细说明,一个ontology往往就是一个正式的词汇表,其核心作用就在于定义某一领域或领域内的专业词汇以及他们之间的关系。这一系列的基本概念如同工程一座大厦的基石,为交流各方提供了一个统一的认识。在这一系列概念的支持下,知识的搜索、积累和共享的效率将大大提高,真正意义上的知识重用和共享也成为可能
目前最常见的就是序列本体论(SO)和基因本体论(GO)。当然还有许多其他的本体论,都列在http://www.obofoundry.org/. 可以去寻找和自己研究领域相关的本体论,比如说植物本体论(PO)用于定义植物基因组数据中的和解剖,形学,生长发育相关的信息。
实际上,生信本质依旧还是序列分析。序列分析比较关注两个内容:
- 这个DNA片段是什么?(注释或分类)
- 这个DNA片段做什么?(功能分析)
de novo 基因组组装就是要构建原来的基因组,然后对上面的片段进行注释,判断是蛋白质还是非编码蛋白。RNA-Seq就是想通过差异表达的转录本来解释表型。RNA-Seq实验的理想结果就是找到DNA功能的机制以及如何产生观察的表型。
序列本体论(Sequence Ontology)
所谓的序列本体论(SO),其实就是定义基因组不同区域的feature。 比如说可以从Sequence Ontology Browser寻找words的定义
例如搜索X_element_combinatorial_repeat就会得到它的定义,还有他的关系图。
X element combinatorial repeat
An X element combinatorial repeat is a repeat region located between the X element and the telomere or adjacent Y' element.
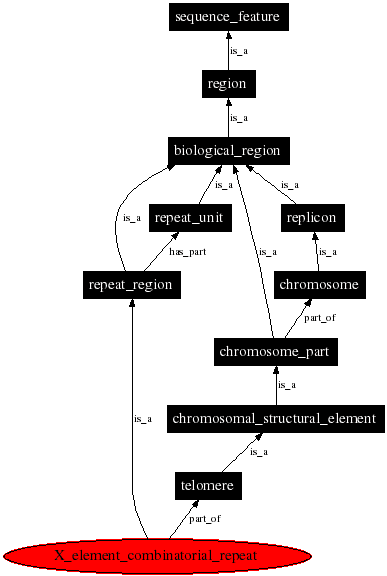
PS: 不是所有序列数据都遵守SO,如CDS的定义。
如何查看SO数据
尽管在日常使用中并不需要接触原始SO数据,但是花点时间理解它是如何组织还是很有必要的
可以在 https://github.com/The-Sequence-Ontology/SO-Ontologies 寻找SO数据。
URL=https://raw.githubusercontent.com/The-Sequence-Ontology/SO-Ontologies/master/so-simple.obo curl $URL > so.obo # 了解有多少term grep 'Term' so.obo | wc -l # 快速查找SO cat so.obo | grep 'PCR' -B 2 -A 2
当你查看GFF文件的时候,如果好奇里面的mRNA, gene的定义就可以用grep进行查找。
Gene Ontology
其实目前最完善的还是基因本体论,也就是GO。基因本体论(GO)是一个受控词汇,用于将每个基因连接到一个或多个功能。
基因本体论用于分类基因产物,而非基因本身。因为同一个基因可以有不同产物,行使不同的功能。
GO必须知道的几个概念
一: GO的组织结构
要记住GO分为三类, CC(细胞组分), MF(分子功能)和BP(生物学过程)。分别回答了基因的产物在哪里发挥功能,如何发挥功能,以及为什么需要该产物这三个问题。
二: GO词条的组织形式
GO本体被构造为有向的非循环图,其中每个术语定义了与同一域中的一个或多个其他术语的关系,并且有时与其他域有关。比如说Golgi Cisterna的GO词条为(GO:0031985),在Quick GO的展示如下
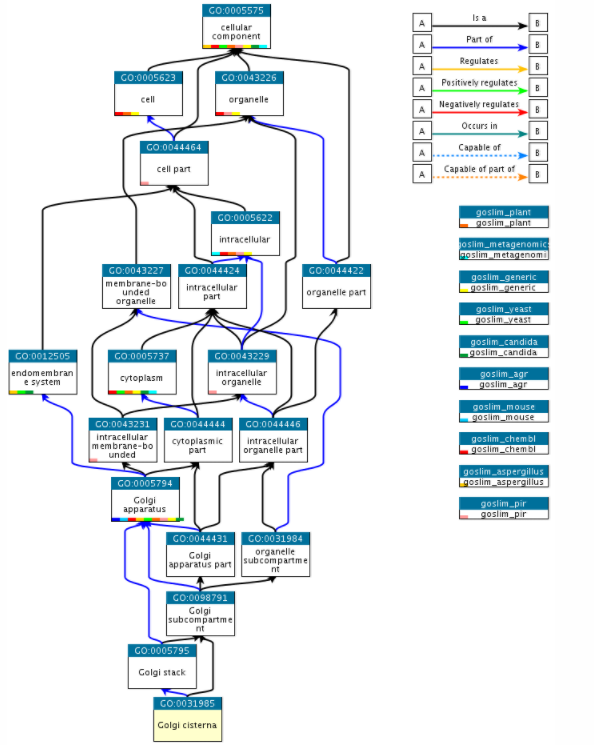
三: GO数据的存储格式
使用GO的时候一般需要GO定义文件和GO关联文件。GO定义文件存放GO词条的定义,而GO关联文件则是不同命名体系与GO词条的映射关系。
实际使用时并不需要获取GO的原始数据,但是能够了解它储存内容的话有助于从多个角度理解生物学。可以从 GO Download数据下载
curl -OL http://purl.obolibrary.org/obo/go.obo
GO词义文件所包含的内容如下:
[Term] # 编号 id: GO:0000002 # 全称 name: mitochondrial genome maintenance # 命名空间,BP, CC OR MF namespace: biological_process # 定义 def: "The maintenance of the structure and integrity of the mitochondrial genome; includes replication and segregation of the mitochondrial chromosome." [GOC:ai, GOC:vw] # 从属关系 is_a: GO:0007005 ! mitochondrion organization
四: GO数据库更新
由于研究的深入,GO的注释数据库会不断地更新。而GO富集分析就和数据库的大小有很大关系。注意了,当公司给你富集分析结果时,一定要注意他们所用的分析工具及其GO数据库版本。
生信数据的功能分析
大部分人学习生物信息学,其实就是为了对数据做出合理的解释,都希望从生物角度获得新的洞见。但是很尴尬,大家花了大部分时间用于收集数据,却对已有的数据的组织和分类及其短视。好像把数据丢到数据库,问题就会自动解决一样。
然而,组织已有的知识是一件非常吃力,但未必讨好的工作。并且大家都指望别人能够搞定这件事情,而不愿意自己动手。于是就导致了”公地悲剧“。一些组织和公司就从中看到了商机,京都基因和基因组百科全书(KEGG)改变授权,对大批量数据检索收费,商业公司构建了专门的数据库进行出售。
因此,商业就是最大的善。它让资源得到合理地配置,让数据变得更有价值,而不是一堆paper。也让科学家们意识上,不合理存放已有数据会有多大的代价。
接下来,让我们聊一块钱的富集分析,其实应该说通路分析(pathway analysis),更好听的说法是功能分析(functional analysis)。也就是当你得到一堆基因或者蛋白后,最常用的方法。目前有三类算法:
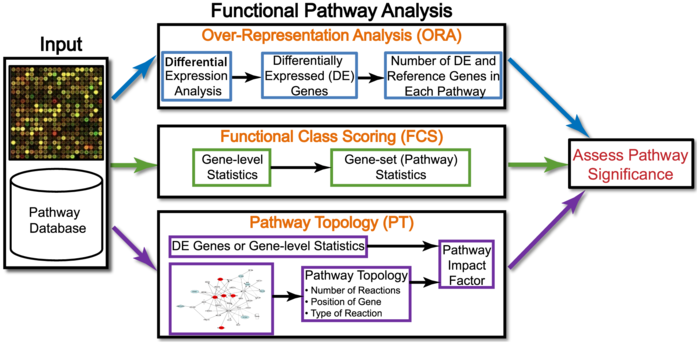
功能分析的算法演化
推荐先去阅读Ten Years of Pathway Analysis: Current Approaches and Outstanding Challenges
第一代:Over-Representation Analysis
ORA翻译成中文就是过表征分析,其实就想看看某类功能或分类和随机事件相比是否有更明显的趋势。这就好比经典统计学中的白球和黑球的抽样问题,如果黑箱中的白球比黑球多,体现在抽样上就是白球会比黑球更容易被抽到。
ORA分析需要你提供4类输入,
- 一共有多少个基因,也就是背景
- 属于某分类的基因有多少个
- 样本一共有多少个基因
- 样本属于某分类的有多少个基因
之后通过超几何分布或2X2独立表进行检验
ORA是公司标准化流程中必备的一步,因为这个方法最简单,当然出现的也足够早。大家总是不太愿意去尝试新鲜事物,不是吗?但是ORA其实是存在很多问题,
- ORA没有考虑到基因的表达水平,仅仅关注基因是否属于分类
- ORA仅仅使用部分数据,存在主观臆断
- 基因和功能被认为是相互独立。这只是一种统计学假设而已,实际情况下并非如此。
但是既然ORA还是目前最常用的方法, 我们还是要尽可能保证这个方法的结果是可靠的。比如说符合如下要求:
- 公司的结题报告中用于富集分析的数据库与时俱进,
- 公司的结题报告中用于富集分析的数据库一定与时俱进,
- 公司的结题报告中用于富集分析的数据库必须与时俱进,
毕竟2016年,Nature Methods 专门写了Impact of outdated gene annotations on pathway enrichment analysis 吐槽大家还在用老旧的DAVID。
第二代:Functional Class Scoring (FCS)
由于ORA方法存在很多弊端,于是就出现了FCS算法。它的基本假设是:虽然单个基因的巨大改变会对通路有显著性印象,但是那些功能相关的类似微效基因累加后也能有显著效果。换句话说,英雄人物可以在某种程度上改变历史进程,但是人民群众的力量也是不容小觑。
实现FCS方法需要三步:
第一步: 通过实验计算出单个基因的基因水平(gene-level)的统计值,比如说基因差异表达衡量会用到的ANOVA,Q-statistic, 信噪比, t-test, Z-score等。
第二步: 同一条通路上所有基因的基因水平(gene-level)统计值聚合成单个通路水平(pathway-level)的统计值。可选方法有,Kolmogorov-Smirnov statistic [21,29],基因水平统计值的和, 均值或中位数, Wilcoxon rank sum, maxmean statistic。
第三步:评估通路水平统计显著性。这一步所需要的统计学思想是重抽样(bootstrap)。也就是对于一个特定通路而言,随机排序和按照一定规则排序是否有差异。
虽然FCS已经比ORA有很大提升了,但实际上依旧有不足。第一,它是单独分析每个通路,而不是多通路组合分析。第二,FCS也只将基因表达的差异用做给定通路的排序而已。比如说A和B的表达量分别改变了2倍和20倍,但是对于不同的通路而言,A和B的排名就有可能相同。
第三代: Pathway Topology (PT)-Based Approaches
为了克服第二代方法, 于是有了第三代基于通路拓扑学的方法。但是这类方法需要用到给定通路互作的信息,而目前相关的数据库不够,于是这个方法只是美妙的设想
富集分析的工具
biostar hanbook里面提到的所有的工具我都不推荐,你也不需要浪费时间去看了。只要知道clusterProfiler就行了,它支持ORA和FCS两类算法。函数为
- enrichGO, gseGO: GO富集分析
- enrichKEGG, gseKEGG: KEGG富集分析
- enrichDAVID: DAVID富集分析
划重点: clusterProfiler的KEGG数据库与时俱进。
这个工具目前唯一的问题就是知道的人还不够。以及大家习惯用网页工具,对代码的无名恐惧,以至于不敢去尝试。还有大家用软件非常害怕去读文档。
推荐阅读