

从埃博拉数据中Call SNPs
# 从多个样品中Call SNPS
# 从埃博拉项目中获取多个数据集。
# Ouch! 数据是以另一个序列作为参考来比对的。
# 那我们准备一个新的参考序列吧,没别的解决方法。
#* NCBI的这套软件最近运行都有问题,建议直接到NCBI下载好再上传服务器。
efetch -id KM034562 -format gb -db nucleotide > KM034562.gb readseq -format=FASTA -o ~/refs/852/KM034562.fa KM034562.gb readseq -format=GFF -o KM034562.gff KM034562.gb bwa index ~/refs/852/KM034562.fa samtools faidx ~/refs/852/KM034562.fa
# 获取运行信息文件
esearch -db sra -query PRJNA257197 | efetch -format runinfo > runinfo.csv
# 选一些SRR(如果觉得冒险的话,可以弄多一些)
cat runinfo.csv | cut -f 1 -d ',' | grep SRR | head -10 > sraids.txt
# 我们把数据放到另一个文件夹中。
# 将会有很多文件。
mkdir -p sra
mkdir -p bam
# 从文件读取每个SRA ID
cat sraids.txt | awk ' { print "fastq-dump -X 20000 --split-files -O sra " $1 } ' > get-data.sh
#* 查看一下get-data.sh的内容
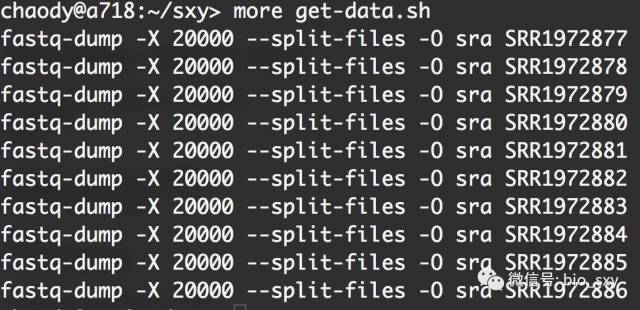
# 这个脚本将会执行数据
#* 数据的下载,以及将格式从sra变为fastq。
#* 解释一下这条命令的意思:fastq-dump -X 20000 --split-files -O sra SRR1972877
#* -X 20000是指拆分获取前20000个spot
#* 如果大家了解fastq的格式的话,一个spot就是指一条完整的测序信息,包含四个组成:名字、序列、+号(+号后面可以加别的说明信息)和测序质量信息。
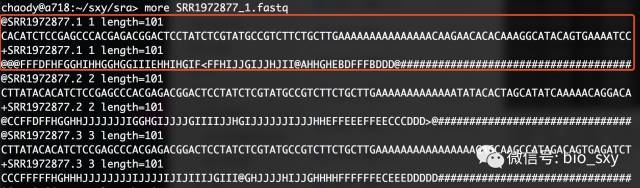
#* --split-files就是把双端测序拆分成两个fastq文件,并且会自动在名字(SRR1972877)后加后缀。
#* -O sra 是指把拆分好的文件放到sra这个文件夹下。
#* 我们可以ls -al查看一下:
bash get-data.sh
# 现在来比对。
# align-ebola 的脚本要先存在。该脚本将比对一个SRA文件并产生一个BAM文件。
#* align-ebola 的脚本在本章节末。
cat sraids.txt | awk ' { print "bash align-ebola.sh " $1 } ' > align-data.sh
bash align-data.sh
# 从所有的数据集中Call snps。
freebayes -p 1 -f ~/refs/852/KM034562.fa bam/*.bam > freebayes.vcf
Alignment script: align-ebola.sh
# 比较两种比对方式的输出。 # 用法: bash align-ebola.sh SRA-RUN-ID # 把比对文件放到一个单独的文件夹。 mkdir -p bam # 在错误处停止运行。 set -ue # 数据文件名。 NAME=$1 # 从传入的参数创建read名字。 # ABC会变为sra/ABC_1.fastq 和 sra/ABC_2.fastq # 形成输入名称。 READ1=sra/"$NAME"_1.fastq READ2=sra/"$NAME"_2.fastq # 参考序列文件。必须两种比对软件都对其进行索引。 REFS=~/refs/852/KM034562.fa # 我们要加一个read group到mapping #* \t表示的是tab分隔 GROUP="@RG\tID:$NAME\tSM:$NAME\tLB:$NAME" # 运行bwa并生成bwa.sam。 # 生成一个排过序的bam文件 # 这将是bam文件的名字。T BAM=bam/$NAME.bam bwa mem -R $GROUP $REFS $READ1 $READ2 2> logfile.txt | samtools view -b - | samtools sort -o - tmp > $BAM echo $? samtools index $BAM echo "*** Created alignment file $BAM" # 打印简单的统计。 samtools flagstat $BAM | grep proper
创建间隔数据
# 获取埃博拉病毒基因组数据和特征(假如你还没有)。
# 一个演示的基因组。也可以是其他的。
efetch -id KM034562 -format gb -db nucleotide > KM034562.gb readseq -format=FASTA -o ~/refs/852/KM034562.fa KM034562.gb readseq -format=GFF -o KM034562.gff KM034562.gb
# 我们将会手动用一个编辑器创建BED和GFF文件,并展示它们。
# BED文件示例, 列是tab分隔的:(当你用浏览器复制粘贴,通常会变为了空格)
KM034562 100 200 Happy 0 -# Example BEDgraph file
KM034562 100 200 50
KM034562 150 250 -50# GTF示例文件
KM034562 lecture CDS 100 200 . - . gene "Happy"; protein_id "HAP2"# GFF示例文件
##gff-版本3
KM034562 lecture CDS 100 200 . - . gene=Happy; protein_id=HAP2# 数据展示:分割示例
# GTF的
##gff-版本2
KM034562 demo exon 1000 2000 . + . gene_id "Happy"; transcript_id "Sad";
KM034562 demo exon 3000 4000 . + . gene_id "Happy"; transcript_id "Sad";
KM034562 demo exon 5000 6000 . + . gene_id "Happy"; transcript_id "Sad";
KM034562 demo exon 7000 8000 . + . gene_id "Happy"; transcript_id "Sad";
KM034562 demo exon 1000 2000 . + . gene_id "Happy"; transcript_id "Mad";
KM034562 demo exon 5000 6000 . + . gene_id "Happy"; transcript_id "Mad";
KM034562 demo exon 7000 8000 . + . gene_id "Happy"; transcript_id "Mad";
KM034562 demo exon 1000 2000 . + . gene_id "Happy"; transcript_id "Bad";
KM034562 demo exon 5000 6000 . + . gene_id "Happy"; transcript_id "Bad";# GFF展示
##gff-版本 3
KM034562 demo exon 1000 2000 . + . Parent=Sad,Mad,Bad;
KM034562 demo exon 3000 4000 . + . Parent=Sad;
KM034562 demo exon 5000 6000 . + . Parent=Sad,Mad,Bad;
KM034562 demo exon 7000 8000 . + . Parent=Sad,Mad;# BED 展示
KM034562 999 8000 Sad 0 + 999 8000 . 4 1000,1000,1000,1000 0,2000,4000,6000
KM034562 999 8000 Mad 0 + 999 8000 . 3 1000,1000,1000 0,4000,6000
KM034562 999 8000 Bad 0 + 999 8000 . 3 1000,1000 0,4000