

随着测序技术的不断发展,宏基因组研究也越来越多。下面整一份从网上收集来的宏基因组研究的资料,与大家分享。为了满足一下不懂宏基因组同学的好奇,顺带从百度百科上取来一份词条作为介绍。
定义
宏基因组 ( Metagenome)(也称微生物环境基因组 Microbial Environmental Genome, 或元基因组) 。是由 Handelsman 等 1998 年提出的新名词, 其定义为“the genomes of the total microbiota found in nature” , 即生境中全部微小生物遗传物质的总和。它包含了可培养的和未可培养的微生物的基因, 目前主要指环境样品中的细菌和真菌的基因组总和。而所谓宏基因组学 (或元基因组学, metagenomics) 就是一种以环境样品中的微生物群体基因组为研究对象, 以功能基因筛选和/或测序分析为研究手段, 以微生物多样性、 种群结构、 进化关系、 功能活性、 相互协作关系及与环境之间的关系为研究目的的新的微生物研究方法。(来自百度百科:http://baike.baidu.com/view/1939711.htm)
测序部分:
宏基因组测序需要提供什么样品要求?
答:(1)提供环境微生物的基因组DNA或者扩增产物,OD值在1.8~2.0 之间;样品浓度大于30 ng/l;每次样品制备需要10μg样品,如果需要多次制备样品,则需要样品总量=制备样μg品次数*10 ug。
(2)DNA样品请置于-20℃保存;请提供DNA样品具体浓度、体积、制备时间、溶剂名称。请同时附上QC数据,包括电泳胶图、分光光度或Nanodrop仪器检测数据。
(3)样品请置于1.5 ml管中,管上注明样品名称、浓度以及制备时间,管口使用Parafilm封口。建议使用干冰运输,并且尽量选用较快的邮递方式,以降低运输过程中样品降解的可能性。
宏基因组测序样品总DNA的提取及基因或基因组DNA的富集注意事项?
答:提取的样品DNA必须可以代表特定环境中微生物的种类,除需严格遵循取样规则外,取样中应尽量避免对样本的干扰,缩短保存和运输的时间,使样品尽可能代表自然状态下的微生物原貌,获得高质量环境样品中的总DNA是宏基因组文库构建的关键之一。要采用合适的方法,既要尽可能地完全抽提出环境样品中的DNA,又要保持较大的片段以获得完整的目的基因或基因簇。所以总的提取总是在最大提取量和最小剪切力之间折中。应严格操作,谨防污染,并且保持DNA 片段的完整和纯度。为了更好地反映环境中的微生物种群并且提高阳性克隆的占有率,需要在克隆之前通过不同的方法对感兴趣的目的基因或基因组进行富集,常用的富集方法有稳定同位素探针、抑制性消减杂交、差异显示、噬菌体展示、 亲和捕获及DNA微阵列等技术。
采用Solexa进行宏基因组测序,测序文库构建方法及质量控制如何?
答:采用Solexa进行宏基因组 DNA测序,首先对特定环境微生物种群全基因组DNA进行提取。在提取微生物种群的DNA后制备DNA文库,具体步骤如下:
(1)将DNA随机打断成200-500bp的片段;
(2)对DNA末端进行修复;
(3)将“A”碱基加入到DNA片段的3’末端;
(4)在DNA片段的末端加上接头;
(5)纯化连接产物;
(6)PCR扩增连上接头的DNA片段;
(7)检测测序文库。
生物信息学分析部分:
下面先放一张宏基因组生物信息学数据分析图的流程图,供大家对着里面的基本过程有一个快速的了解。
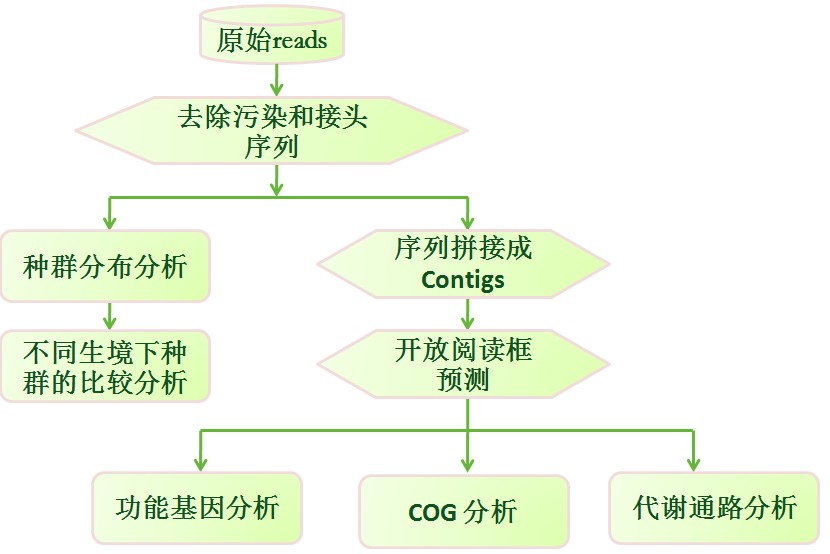
如何估计宏基因组样本中的物种组成及丰度?
答:宏基因组中的物种分类,一般用OTU (operational taxonomic unit), 即可操作物种单元,来表示。在典型情况下,原核生物的OUT使用16S rDNA来衡量,真核生物的OUT使用18s rDNA来衡量。
但选择16S/18S rDNA鉴定物种,存在以下几个问题:1)rDNA之间的平行转移来干扰rDNA鉴定的可靠性。2)在单个细菌中,16r DNA可能存在序列不同的几个拷贝,干扰估计OTU数目的准确性。所以,其他备选的标记基因,比如单拷贝的看家基因被推荐用来作为菌种鉴定的标记。
如何衡量样本中物种的多样性?
答: 为了估算测序的物种的比例,通常用rarefaction curse来表示。

宏基因组如何做De Novo拼接?
答:由于宏基因组测序的覆盖率通常是不完全的,所以组装所需要的序列并不是很完整。并且组装的时候,可能会把来自不同分类单元(OTU)的序列组装在一起,产生嵌合体基因组。Phrap,Forge,Arachne,JAZZ和Celera Assembler等可用来组装由sanger法产生的宏基因组序列。这些算法大部分都利用mate-pair信息来参与组装。这些算法用顶点来代表每条read,互相重叠的read之间用边连起来,它们的组装问题可以转换成“哈密尔顿路径”搜索问题,即找到一条路径走过所有顶点,且每个顶点只走一次。
如何进行菌群间差异分析?
答:有几种基于序列特征的比较,包括样品间GC含量的比较,微生物基因组大小的比较,系统发育关系树的比较和功能组分的比较。许多比较分析都用到了关联统计学的方法,通常假设有几种元数据影响观测到的宏基因组群体的组分。主成分分析(PCA)和非度量多维标度(NM-MDS)用来图形化展示数据并揭示有哪些因素最影响数据。
有几种进行宏基因组比较分析的软件。第一个是MEGAN, 可以比较两个或几个标准化后的样品的GC含量。第二种是MG-RAST,提供了一种比较功能和基于序列的分析来上传样本。第三种是CAMERA,提供了BLAST接口让客户可以比对40多种现有的宏基因组数据。
如何预测编码基因?
答:目前发现编码基因的方法有两种。一种是基于BLAST比对的方法,这种方法通过比对已有的数据库,可以发现宏基因组数据中有哪些已知基因的同源基因的存在,但缺陷是找不到哪些和已经基因没有同源关系的新基因。第二方法是重新预测基因的方法,这些方法大部分是基于有指导学习和统计模式识别的方法,包括隐马尔科夫模型。GeneMark.hmm就是基于单密码子频率的非均一马尔科夫模型来预测基因的软件,当这些软件用到宏基因组数据上时,这些软件通常无法确定部分的ORF,即使这些 ORF是真实基因的一部分。
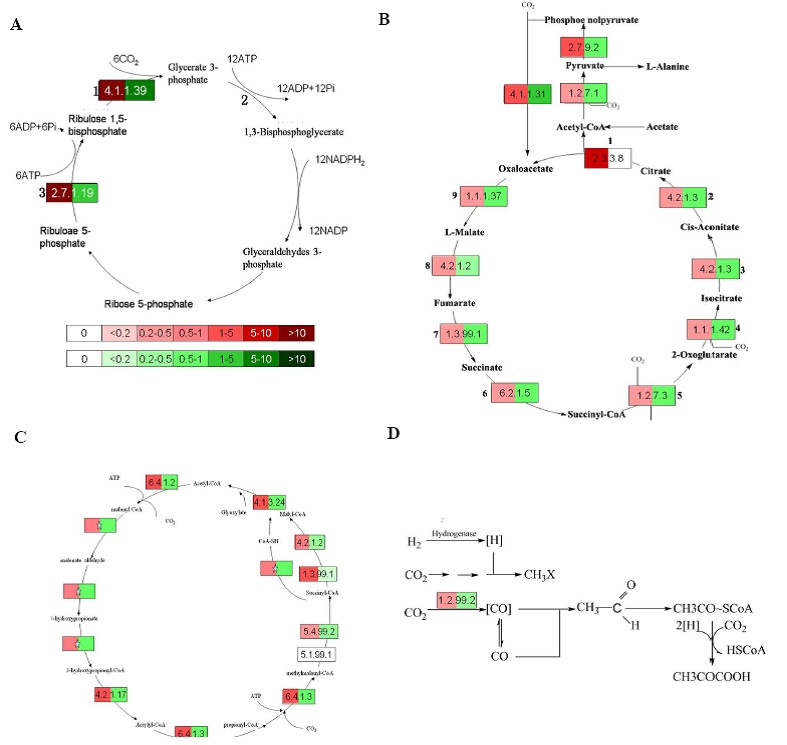
如何进行代谢通路分析(pathway analysis)?
答: 代谢通路分析是为了研究某一个环境中各种代谢途径的富集程度。一般需要根据统计检验方法(如P-value)来筛选。常用的代谢通路数据库有KEGG、Reactome、BioCyc、RegulonDB、 WikiPathwans等。
KEGG(Kyoto Encyclopedia of Genes and Genomes)是系统分析基因功能、基因组信息数据库,它有助于研究者把基因及表达信息作为一个整体网络进行研究。基因组信息存储在GENES数据库中,包括完整和部分测序的基因组序列;更高级的功能信息存储在Pathway数据库里,包括图解的细胞生化过程如代谢、膜转运、信号传递、细胞周期,还包括同系保守的子通路等信息。KEGG的另一个数据库是LIGAND,包括关于化学物质、酶分子、酶反应等信息,可以免费获取。KEGG提供的整合代谢途径 (Pathway)查询十分出色,包括碳水化合物、核苷、氨基酸等的代谢及有机物的生物降解,不仅提供了所有可能的代谢途径,而且对催化各步反应的酶进行了全面的注解,包含有氨基酸序列、PDB的链接等。
本文中问答部分内容摘选自:http://www.igenomics.com.cn,仅供大家参考。欢迎大家补充。如有疑问,请到问答社区交流(http://home.plob.org)
1F
楼主威武
2F
狂顶