

单细胞转录组、蛋白组、表观组学等单细胞技术的发展为研究细胞周期、细胞分化等细胞动态过程提供了新的机会。使用轨迹推断(TI,trajectory inference)的方法可以根据测序的细胞之间表达模式的相似性对单细胞沿着轨迹进行排序,以此来模拟细胞动态变化的过程。轨迹推断也常被称作“伪时间分析”(pseudotime analysis)。 在过去几年中,已经开发了大量的轨迹推断(后续简称TI)方法,在以下几个收录单细胞工具的库中,TI分析的工具是最大的类别之一。
单细胞领域的新用户面临着大量的TI方法选择,而没有一个明确的想法可以最佳地解决他们的问题,纵然大多数人选择了引用率比较高的Monocle2 (Qiu et al., 2017)做TI分析,但是Monocle2也常常不能做出令人满意的TI结果,现实中很多人往往选择筛选细胞减少计算量以应对大数据集的项目。不同的数据集可能对应不同的轨迹模型(线性的、分叉、树形、循环图、不连续图),繁多的TI方法在性能、可扩展性、健壮性和可用性上存在差异,各有其优缺点。
2019年发表在
Nature biotechnology的一篇文章A comparison of single-cell trajectory inference methods(Saelens et al., 2019)对45种TI方法在110个真实数据集和229个合成数据集中进行了全面比较,主要评估了TI方法的准确性、可扩展性、稳定性和可用性四个方面
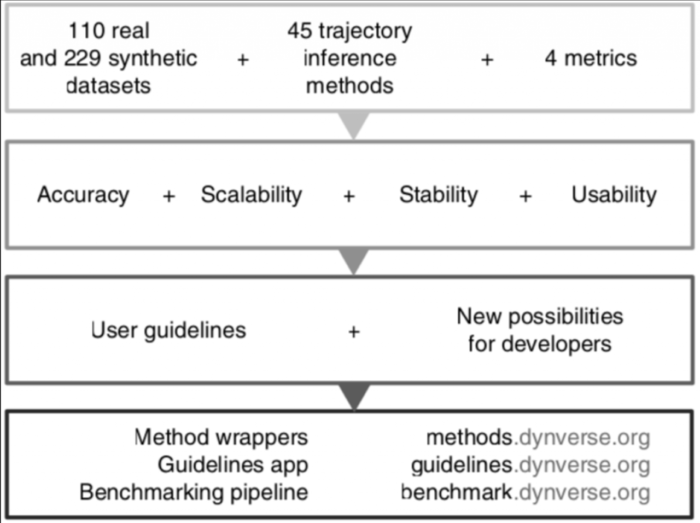
以下是文章(Saelens et al., 2019)的主要可参考点:
- [x] 文章的研究评估发现:发现当前TI方法之间存在很大的互补性,数据集的特性不同,执行效果最佳的TI方法也不同
- [x] 作者基于评估的结果,制定了一套准则,以帮助用户为自己的数据集选择最佳方法
http://guidelines.dynverse.org - [x] 开发了一个R包--dyno,把文章中测试过的TI方法进行了封装,目前已封装了55种,允许用户根据
guidelines.dynverse推荐的方法,自由选择进行TI分析,dyno采用统一的输入,同时也对不同TI方法的输出做了统一
不得不说dyno真的很吸引人,一个R包就解决了那么多TI包的安装和使用问题,可以让用户在自己项目的数据集上方便的尝试多种TI方法,所有TI方法的输入统一成一种格式,提供了统一的可视化模式,方便用户比较不同TI方法在自己项目数据集上的表现
不同TI方法结果的比较策略
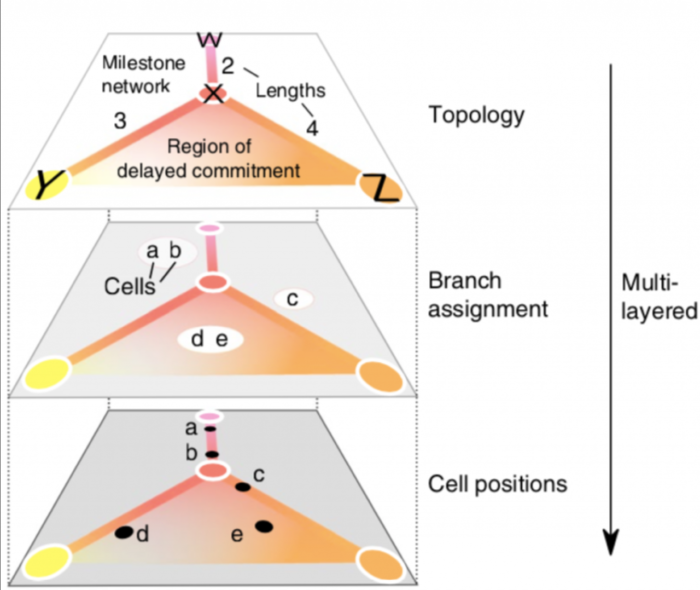
为了使不同TI方法的输出彼此直接可比,作者开发了一个通用的概率模型来表示来自所有可能来源的轨迹,如上图所示。在这个模型中。
一 整体拓扑结构由里程碑(milestones)网络表示
细胞被放置在每组相互连接的里程碑所形成的空间内。
二 不同TI结果标准化归类输出
几乎每种TI方法都返回一个唯一的轨迹结果输出,作者总结了TI方法的结果,把这些结果分为7个不同的公共轨迹模型:
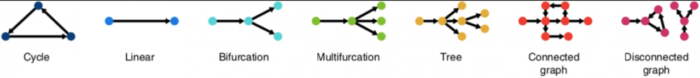
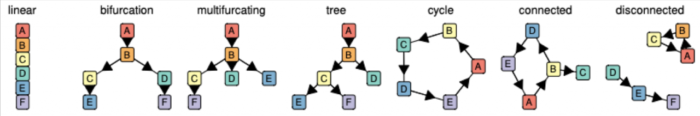
对每一种TI方法的结果做归类,假如一组数据用某一种TI的结果被归为了Branch assignment这个类别,就把这种TI方法的分析结果转化成统一的Branch assignment类标准的公共轨迹模型输出。 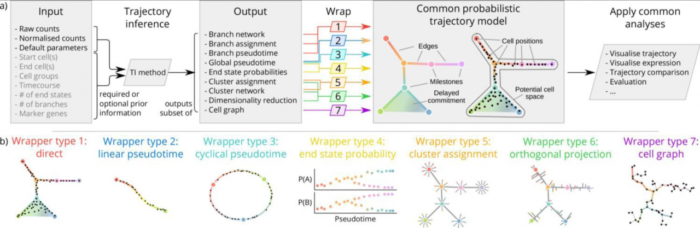
这个通用模型可以对任何TI方法产生的轨迹模型执行通用分析功能,例如轨迹的可视化及与黄金数据集结果的比较。
三 不同TI方法归类
TI方法之间最大区别在于,是否固定拓扑,作者定义了7种可能的拓扑类型,从最基本的拓扑(线性,循环和分叉)到更复杂的拓扑(连通图和非连通图)。大多数TI方法要么着重于推断线性轨迹,要么将搜索范围限制在树或较不复杂的拓扑中,只有少数尝试推断循环或不连续的拓扑),不同TI方法的轨迹模型分类如下图所示:
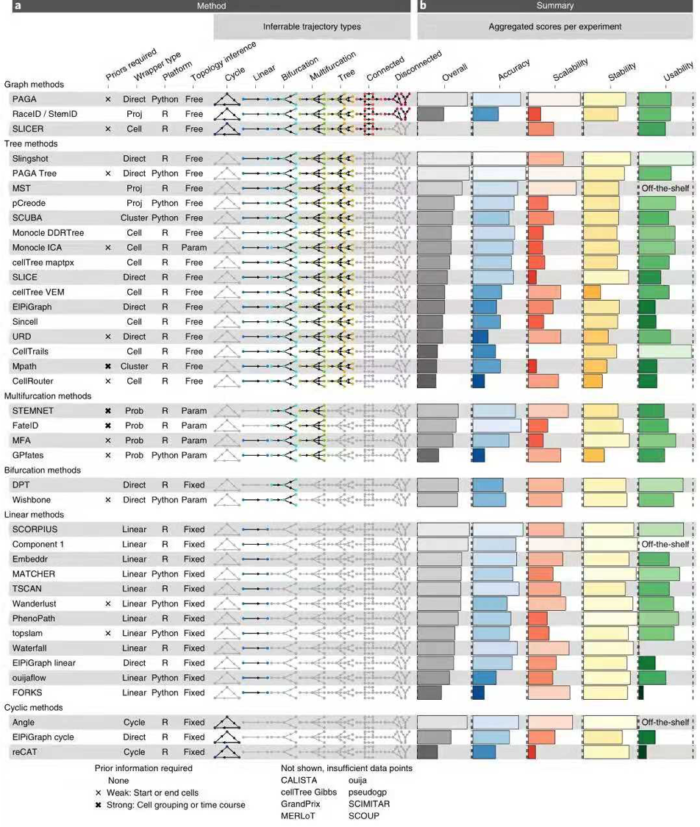
作者从四个核心方面评估了每种方法:
- 在110个真实数据集和229个合成数据集上给出金或银标准的情况下,预测的准确性;
- 关于细胞和特征(例如基因)数量的可扩展性;
- 对数据集进行二次采样后预测的稳定性;
- 工具在软件、文档和手稿方面的可用性。
总体而言,作者发现大多数TI方法在这四个评估标准之间存在很大差异,只有少数方法(例如PAGA,Slingshot和SCORPIUS)比较均衡。
四个核心方面评估

准确性评估(Accuracy)
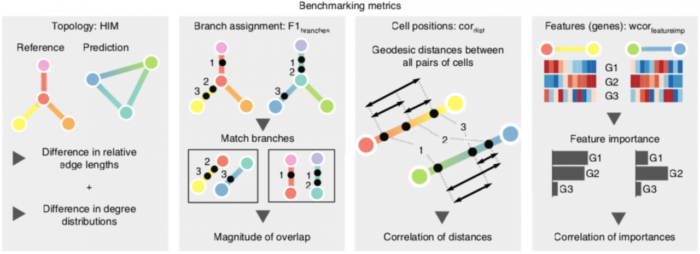
为了将TI方法的结果与先验的参考轨迹进行比较,作者定义了几个度量标准:
metric
- HIM score:考虑了边长和度分布(度-degree 是指网络/图中一个点的与其他点的连接数量,度分布-Degree Distribution 就是整个网络中,各个点的度数量的概率分布)的差异,评估了两种拓扑之间的相似性
- F1 Branches assesses:将细胞分配到分支的相似性
- Cell positions:通过计算成对测地距离之间的相关性,来量化两个轨迹之间的细胞位置相似性
- Features (genes):特征量化了从已知轨迹到预测轨迹的轨迹差异表达特征之间的一致性
dataset source
测试数据由以下两部分构成: * 229个合成数据集:提供最精确参考轨迹 * 110个真实数据集:提供最高生物学相关性
110个真实的数据集来自各种单细胞技术,各种生物体和动态过程,并包含几种类型的拓扑轨迹。 作者把做测试用的真实数据集做了两个分类: * Gold standard:参考轨迹是通过细胞分选或细胞混合而来,不是从表达数据本身中提取 * Silver standard:gold standard之外的数据集
作者使用4个不同的合成数据模拟器合成了229个合成数据集,对于每种模拟,作者都使用一个真实的数据集作为参考,以匹配其尺寸,差异表达基因的数量,丢弃率和其他统计特性: • dyngen:用来模拟细胞调控网络github.com/dynverse/dyngen • dyntoy:缩减空间中表达的随机梯度github.com/dynverse/dyntoy • PROSSTT:从线性模型中抽取表达式,该模型取决于伪时间 • Splatter:模拟不同表达状态之间的非线性路径
trajectory type
作者发现TI方法性能在各个数据集之间的表现变化很大,这表明没有一种万金油的方法适用于每个数据集,即便是PAGA、RaceID/StemID、SLICER这些可以检测大多数轨迹类型。
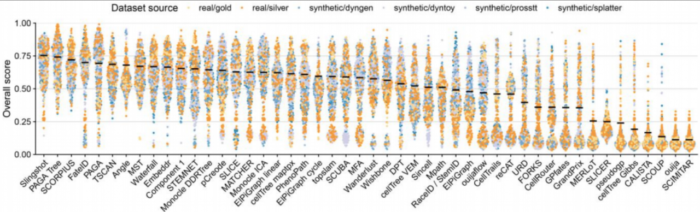
不同数据集来源之间的总体得分与包含金标准的真实数据集的得分具有中等至高度相关性(斯皮尔曼等级相关性在0.5-0.9之间),从而确认了金标准轨迹的准确性以及合成数据的相关性。
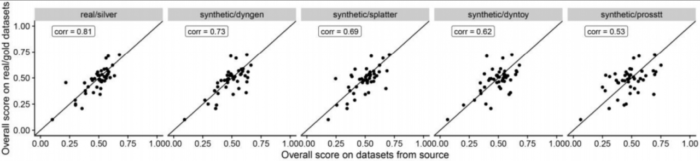
不同的指标(metric)经常彼此不一致,Monocle和PAGA Tree在拓扑分数上得分更高,而其他方法(例如Slingshot)则在细胞排序并将它们放入正确的分支方面更好。 TI方法的性能在很大程度上取决于数据中存在的轨迹类型,Slingshot通常在包含更简单拓扑的数据集上表现更好,PAGA,pCreode和RaceID/StemID在具有树状或更复杂轨迹的数据集上得分更高
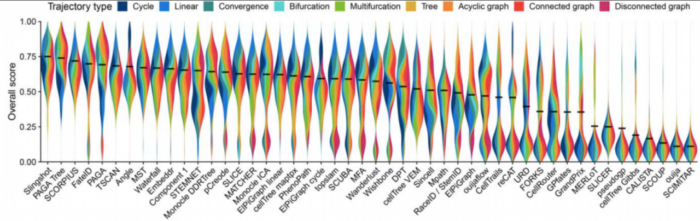
这种情况反映在每种方法检测到的拓扑类型中,因为Slingshot预测的拓扑倾向于包含较少的分支,而PAGA,pCreode和Monocle DDRTree检测到的拓扑倾向于更复杂的拓扑。
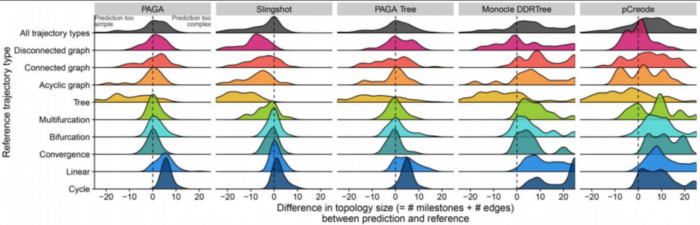
因此,这些分析表明,对于大多数TI方法而言,检测正确的拓扑仍然是一项艰巨的任务,因为就数据中拓扑的复杂性而言,目前的TI方法往往过于乐观或过于悲观。
TI方法之间的互补性
数据集之间的高度可变性以及不同TI方法检测到的拓扑结构的多样性可能表明不同TI方法之间存在一定的互补性,为了测试这一点,作者计算了仅使用所有TI方法的子集时获得顶级模型的可能性,顶级模型被定义为获得的总得分高于最优模型得分的95%。
在所有数据集上,只使用一种TI方法(PAGA Tree)的情况下,有27%可能性获得顶级模型,通过增加其他6种方法(SCORPLUS|Singshot|Angle|Monocle ICA|PAGA|) 以上获得顶级模型的方法组合是一组相对多样化的方法,其中包括严格的线性或循环方法,以及具有广泛轨迹类型范围的方法,例如PAGA,在仅包含线性,分叉或多分支轨迹的数据上,作者发现顶级方法之间具有相似的互补性迹象。
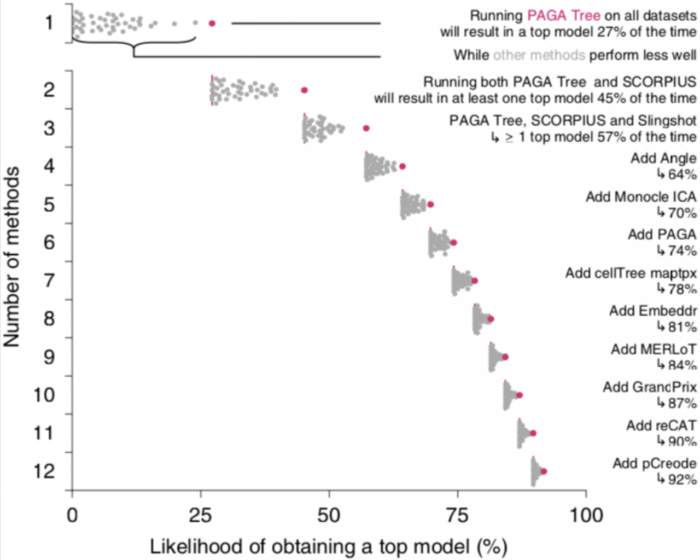
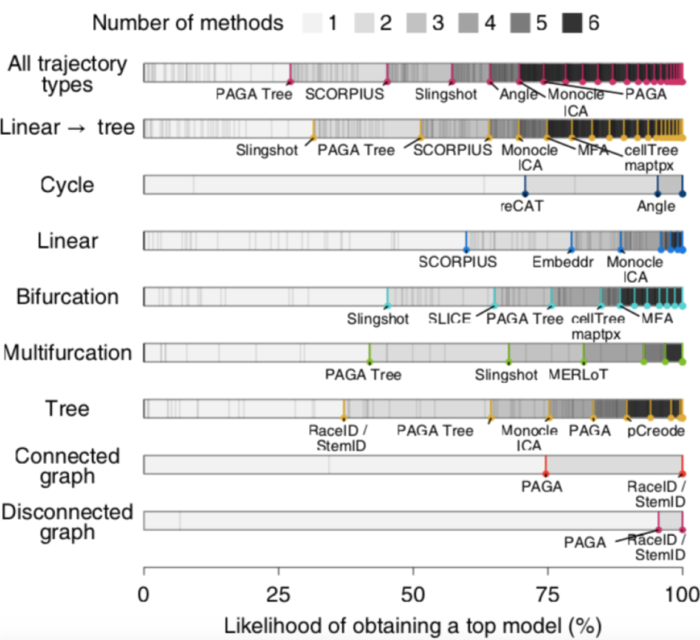
尽管在一个给定数据集的情况下,以上案例中能够用比较少的TI方法获得顶级模型,总体而言,这表明不同方法之间存在相当大的互补性,用户应在其数据上尝试各种TI方法,尤其是在先验知识不清楚的情况下。此外,这也为利用这种互补性构建新的集合方法提供了可能性。
可扩展性评估(Scalability)
早期TI方法构建时的测试数据集在1000个细胞左右,但是现在随着10X等高通量单细胞技术的普及,这些TI方法经常需要处理几万,也许在未来有处理上千万细胞的需求,而且随着单细胞多组学技术的发展(10X ATAC等),每个细胞的需要处理的特征(gene|peak等)也越来越多。所以作者评估了目前的TI方法在处理细胞数、特征数(gene)性能的扩展。
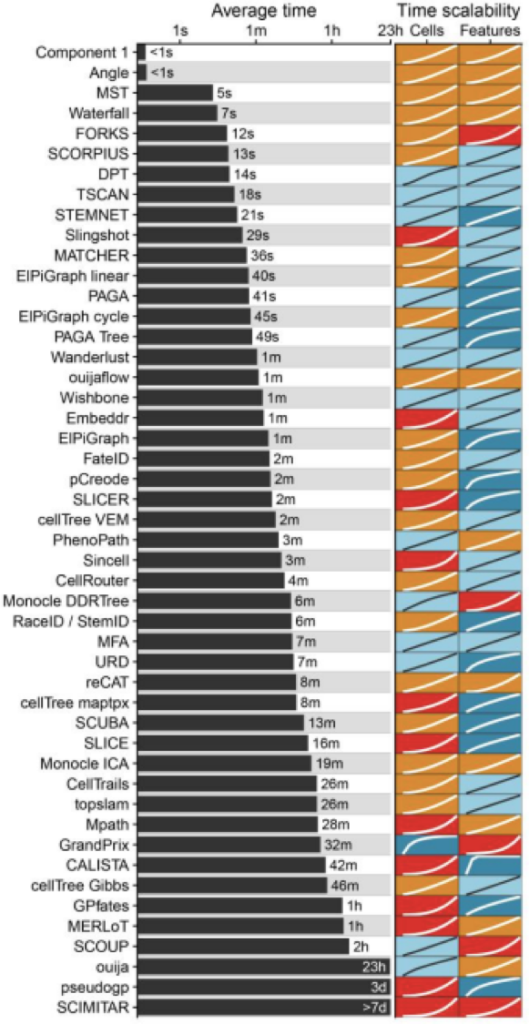
扩展对运行时间的影响
作者发现,大多数TI方法的可扩展性总体上很差,大多数图和树方法无法在一小时内在具有10k个细胞和几千个特征(gene)的数据集上完成,这是典型的10X等基于液滴的单细胞数据集大小。
随着细胞数量的增加,运行时间进一步增加,只有少数几个方法(PAGA, PAGA Tree, Monocle DDRTree, Stemnet and GrandPrix)可以在1天内处理完100万细胞的分析。当处理大量特征的数据集时,某些方法(例如Monocle DDRTree和GrandPrix)也会遇到运行时间不令人满意的情况。
运行时间短的TI方法通常具有两方面特征: 1. 相对于细胞/特征,它们具有线性的时间复杂度 2. 添加新的细胞/特征导致时间增加相对较低
作者发现,在所有方法中,有超过一半的方法具有相对于细胞数量的二次或超二次复杂度,这将使得很难在合理的时间范围内将这些方法中的任何一种应用于细胞量超过1000数据集
![]()
扩展对运行内存的影响
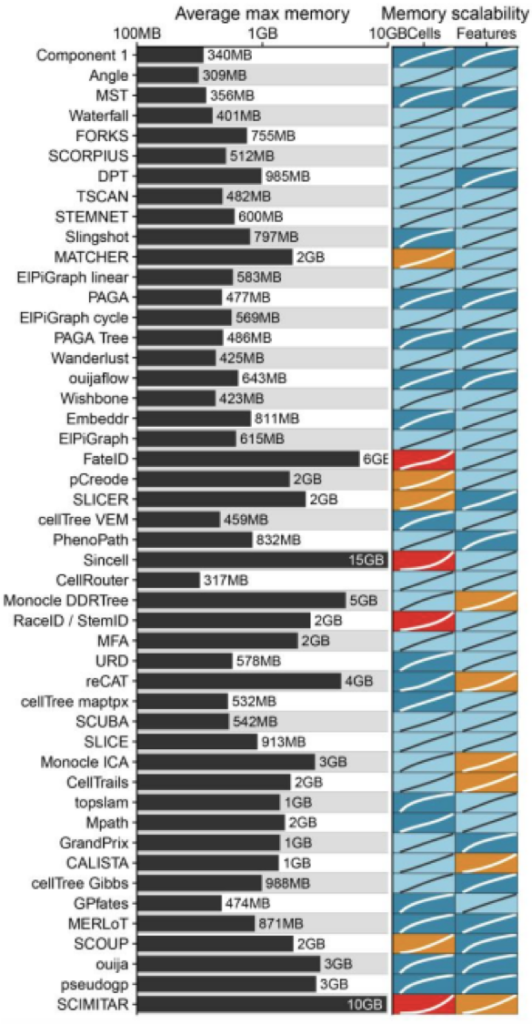
大多数TI方法都具有合理的内存消耗。但是,对于细胞数比较多的数据集而言有一些方法(RaceID / StemID,pCreode和MATCHER)内存需求非常高,对于Monocle DDRTree, SLICE 和 MFA来说,特征值比较多时会消耗比较大的内存。
总的来说数据集的大小是选择合适方法的重要因素,在TI方法开发的时候应该更加注意保持合理的运行时间和内存使用率。
稳定性评估(Stability)
TI方法不仅要能够在合理的时间范围内推断出准确的模型,而且要在给定非常相似的输入数据时生成相似的模型。 为了测试每种方法的稳定性,作者对10个数据集的子集(95%细胞,95%特征)测试了每种方法,并评估每对模型之间的平均相似性和轨迹的准确性。 考虑到通过算法或通过参数固定拓扑的方法的轨迹已经受到很大的限制,因此可以预料,这种方法会产生非常稳定的结果。但是,某些固定拓扑方法仍然产生稍微更稳定的结果,例如线性方法的SCORPIUS和MATCHER以及多分支方法的MFA。 在具有自由拓扑的方法之间,稳定性更加多样化。Slingshot产生的模型比PAGA(树)更稳定,而PAGA(树)又比pCreode和Monocle DDRTree产生更稳定的结果。
可用性评估(Usability)
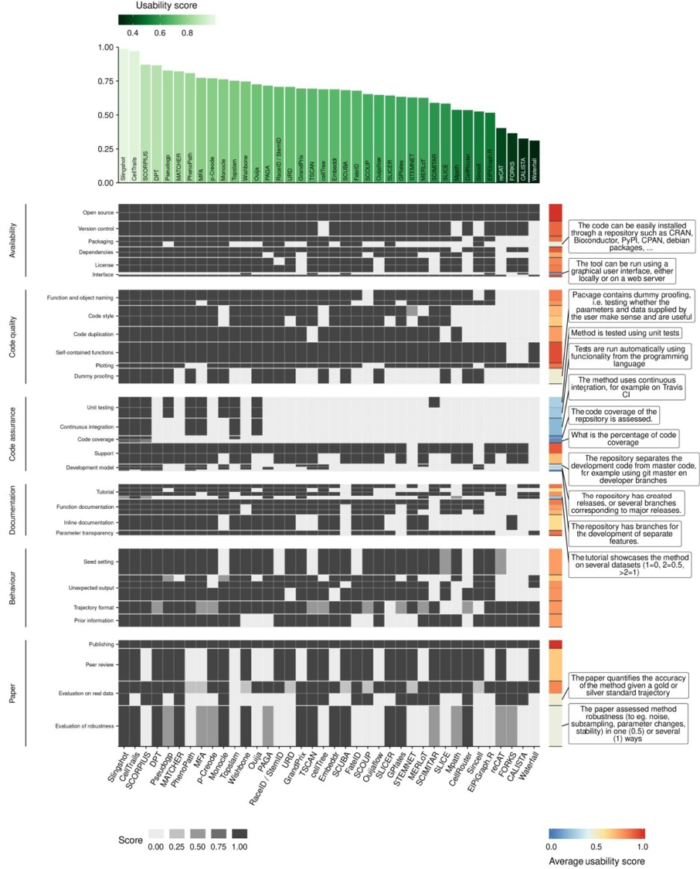
尽管与推断的轨迹的准确性没有直接关系,但一个TI方法能不能被评估实施以及对生物学用户的友好程度也很重要。 作者对每种方法的软件包装、文档、自动代码测试以及发布的期刊做了评估。作者发现大多数方法都满足基本标准,例如教程的可用性和基本代码质量标准,新方法的质量得分比旧方法好一些,
以下几个方面几乎所有的TI方法在某些方面多少有些不足 * Availability * Behaviour * Code assurance * Code quality * Documentation * Paper
只有两种方法具有近乎完美的可用性评分(Slingshot和Celltrails)它们可以用作未来新方法开发的参考。
TI方法选择指导原则
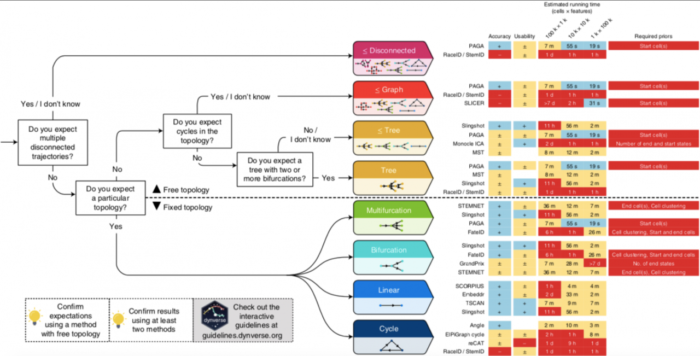
根据基准测试的结果,作者为用户提供了一套TI方法选择实用的指导原则Guidelines.dynverse.org 作者基于这样的假设:由于一种方法的性能很大程度上取决于所研究的轨迹类型,因此方法的选择应主要由数据中预期的轨迹拓扑决定。在大多数情况下,用户可能对预期的轨迹了解得很少,除非期望自己的数据包含固定的轨迹预期,例如多个断开的轨迹、循环或复杂的树形结构。对应每一种预期的轨迹类型Guidelines.dynverse.org都提出了一组不同的最佳方法。数据集的大小和可用的先验信息也会影响方法的选择。这些因素以及其他几个因素都可以在交互式应用程序(guidelines.dynverse.org)中进行动态探索。这个shiny应用程序还可用于查询评估结果,例如过滤数据集或更改评估指标对最终排名的重要性。 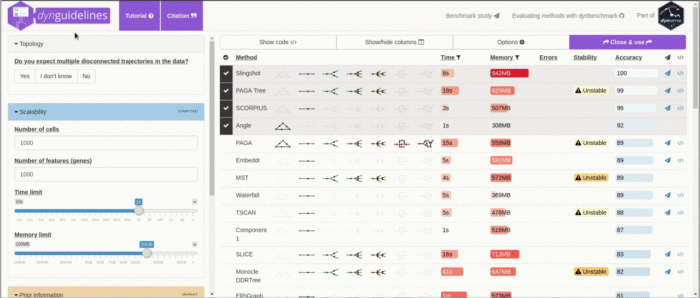
在数据集上尝试不同TI方法的意义
在推断感兴趣的数据集的轨迹时,必须考虑另外两点:
- 轨迹以及其下游结果/假设需要在多种TI方法上得到重现,这样做可以确保轨迹的预测不会是由于给定的参数设置或TI方法所基于的特定算法而产出的偏好性的结果。
- 即使我们知道了预期的轨迹拓扑,也可以尝试一些分支较少的轨迹拓扑假设。如果使用这种方法确认了预期的拓扑,那么它将为用户提供证据支持;当产生更复杂的拓扑时,这可能表明基础生物学比用户预期的要复杂得多。
TI方法输入和输出接口的标准化
TI方法输入和输出接口的标准化对于TI方法的广泛应用非常关键,如果能够实现,那么用户就可以非常轻松方便地在感兴趣的数据集上执行不同的TI方法,以便进行比较预测轨迹并应用下游分析,例如寻找对轨迹或网络推断重要的基因或寻找基因模块集合。
使用作者提供的框架,仅需使用几行R代码(https://methods.dynverse.org)就可重新创建图形。在未来,这个框架可以扩展以兼容其他输入数据,例如空间位置和RNA velocity信息,并简化下游分析。
作者呼吁:需要在TI领域内进行进一步讨论以达成关于轨迹模型的通用接口的共识,其中可能包括不确定性(uncertainty)和基因重要性等其他特征。
TI领域的持续挑战
轨迹推断的领域正在成熟,主要是线性和分叉的轨迹。
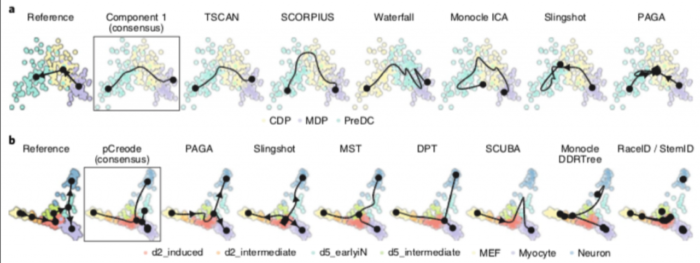
但是在TI成为分析具有复杂轨迹的单细胞组学数据集的可靠工具之前,应解决这些挑战。
- 新方法应该集中在改善树,循环图和不连续拓扑的无偏推论上,因为作者发现:即使使用降维方法可以轻松地确定轨迹方法,大多方法却反复高估或低估基础拓扑的复杂性。
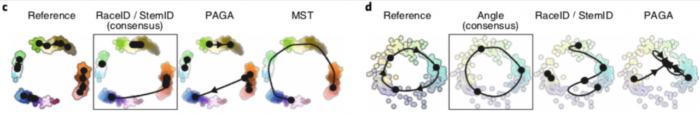
- 更高的代码保证和文档标准可以帮助在单细胞组学领域推广这些工具。
- 应该设计新的工具以随着细胞和特征数量的增加而很好地扩展。作者发现,目前只有少数几种方法可以在合理的时间内处理超过10,000个细胞的数据集。
为了支持这些新工具的开发,作者在https://benchmark.dynverse.org上提供了一系列有关如何包装和评估一种针对本研究中提出的不同措施的方法的方法。
作者的总结
作者通过测试发现一种TI方法的性能在数据集之间可能非常不同,因此在评估中包含了大量的真实和合成数据,从而对不同方法进行了全面的排名。但是目前万金油的方法,TI领域仍然值得继续探索并做出非常有价值的贡献,特别是如果研究者利用新颖的算法,返回更具可扩展性的解决方案或在特定用例中提供独特的见解。作者对不同TI方法互补性的分析也支持了这一点,例如 * PhenoPath:可以在其模型中包含其他协变量 * ouija:可以返回轨迹中每个细胞位置的不确定性度量 * StemID:可以推断出轨迹内分支的方向性
后记
作者对几十种TI方法的输入输出接口进行了标准化,并提供的统一的工具包dyno,下期我们将带来dyno代码实操教学,让你实现安装一个软件方便的体验多种轨迹分析方法。
参考
- Qiu X, Mao Q, Tang Y, et al. Reversed graph embedding resolves complex single-cell trajectories[J]. Nature methods, 2017, 14(10): 979.
- Saelens W, Cannoodt R, Todorov H, et al. A comparison of single-cell trajectory inference methods[J]. Nature biotechnology, 2019, 37(5): 547.