

原文链接:
- https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6072887
摘要
单细胞RNA测序技术的发展加深了我们对于细胞作为功能单元的理解,不仅能基于成百到成千上万个单细胞的基因表达谱得到新的结论,还能发现新的具有特异基因表达谱的细胞群(这在传统转录组测序数据中是很难发现的)。
但是合适的分析和使用单细胞RNA测序后的大量数据并不容易,需要了解从准备细胞到获得合理的结果之间采用的实验和分析方式。
在本篇综述中,作者讨论了这些新技术的基本原理,重点强调单细胞转录组分析中的重要概念。具体地说就是总结了单细胞过滤的质控方法、由于mRNA捕获率低需要进行的标准化和归一化方法和用于降维数据绘制二维图的聚类和可视化的算法。
分析流程
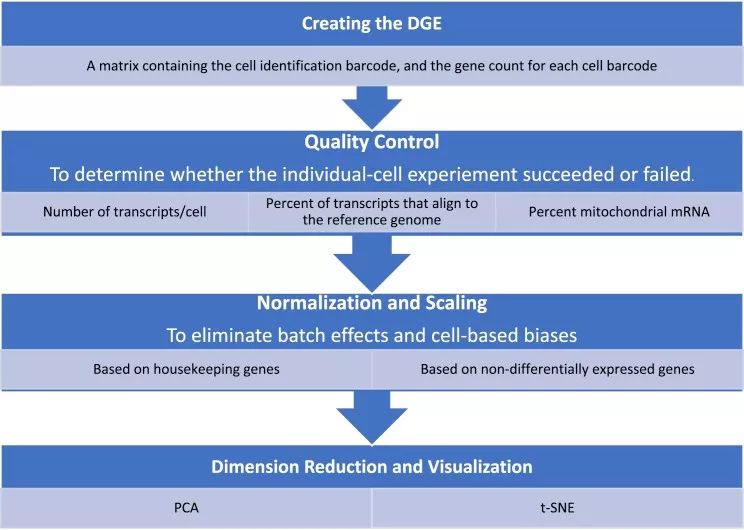
图示:对得到的DGE数据进行质控→依据转录本和细胞数、转录本比对到参考基因组的比例和线粒体mRNA的比例对数据进行质控→用标准化和归一化来除去批次效应和误差→降维和可视化。
正文
早期,单细胞基因表达图谱用于研究少数单个细胞里面特定选择的一些转录本。后来,高通量测序技术和高效细胞分离方法的发展促进了现代单细胞测序平台(例如Fluidigm C1,DropSeq,Chromium 10X,SCI-Seq以及过去十年中开发的许多其他单细胞测序技术)的发展。
表格展示了现在流行的单细胞测序方法,这些方法都依赖于细胞解离技术。价格很大程度上取决于测序细胞的数量、测序深度以及测序平台。表中的价格是单细胞文库制备价格区间的最低价。
| Sequencing Method | Starting Cell No. | Cell Separation | Notes | Cell Capture | Transcript Capture | Representative Library Prep Cost per Cell1 |
|---|---|---|---|---|---|---|
| Fluidigm C12 | ~1,000 cells | cells capture insize-specific chambers | must know the size of cells of interest; allows forstaining and imaging prior to cell rupture | 96- or 800-chamber units areavailable | an average of 6,606 genes/cell (no data on percentage) | $1.70 |
| DropSeq | ~150,000 cells/run | droplet-based separation | remains the mostcost-effective and most customizable | ~5% of cells per run (approximately 7,000 cells) | ~10.7% of the cell’s transcripts | $0.06 |
| Chromium 10X | ~1,700 cells/run | droplet-based separation | the most commerciallysuccessful method; almost fully automated | ~65% of cells per run (approximately 1,000 cells) | ~14% of the cell’s transcripts | $0.10 |
| SCI-Seq | ~500,000 cells (depends on experimental design) | FACS sorter; cellsare never singly isolated | combinatorial indexing of individual methanol-fixedpermeable cells | 5%–10% of cells | ~10%–15% of the cell’s transcripts | $0.05–$0.143 |
表1
[1]: 截至2018年7月
[2]: 基于800-隔离室的中等大小分离平台
[3]: 取决于要测的细胞数和允许的doublets数。
这些技术能够一次检测到数百至成千上万个单细胞的转录组数据。在逆转录或后续步骤中,这些方法都是使用DNA barcode标记mRNA,来判断转录本来自于哪个细胞。尽管每种技术在分离细胞和标记mRNA方面处理方式都不一样,但是它们都使用相似的计算流程处理单细胞转录组数据。
在本篇综述中,作者讨论了流程中最常使用的一些算法,并以DropSeq为主要示例,因为它是性价比最高并且使用最广泛的单细胞基因表达平台(表1)。不过这些方法也同样适用于绝大多数使用DNA barcode 来标记mRNA来源的单细胞测序技术。
单细胞转录组主要应用于:
- 深入了解组织中的细胞异质性。
- 鉴定未知的细胞类型。
- 鉴定已知细胞类型的亚型,原理是在感兴趣的细胞群中寻找差异基因表达模式。
- 从稀有细胞群中分离出信号,这些信号在普通转录组中很难被分离出来。
- 给未知Maker的细胞类型推断可能的Maker,如细胞表面蛋白等。这个原理是由于单细胞转录组分析中会根据细胞间差异表达的基因进行聚类,这样就可以把对聚类影响最大的基因认为是感兴趣细胞群可能的Maker。
- 细胞谱系和分化调控的研究,例如可以诱导一组干细胞分化,并在不同时间点进行单细胞测序可得到分化各个阶段的“snapshot”。这些
snapshot可用于推断细胞到达终末分化状态所遵循的轨迹和在每个分支点受到差异调控的关键基因。
不过这些应用几乎都依赖于由顶尖生物信息学实验室开发的并释放的一些特定的算法。而在本篇综述中,作者重点介绍了用这些特定算法之前必须做的——对数据进行质量控制和标准化,并讨论了简单的细胞聚类和可视化的算法。
基于Droplet方法生成单细胞基因表达数据集
DropSeq和商用的10X都是基于液滴(Droplet)的方法来产生单细胞基因表达数据集。基于液滴型方法是使用微流体芯片将单个细胞与单个凝珠(beads)包裹进油囊化液滴中,理想情况下每个液滴最多包含一个细胞。
凝珠 (beads)上附有数目极多的DNA寡核苷酸探针(DNA oligos)。DNA寡核苷酸的3’末端是一个poly(T)尾巴,可用于捕获细胞中的mRNA (生信宝典注:更准确的说是捕获细胞中有poly-A尾巴的RNA,既有mRNA,也有ncRNA);5’末端是用于标记细胞的cell barcode序列,一个凝珠上结合的所有寡核苷酸的cell barcode都一样;中间还有一个具有高度多样性的唯一分子标识符(unique molecular identifier,UMI),磁珠上的每个寡核苷酸的UMI都不一样 (生信宝典注:这个不能同意,UMI的种类是少于一个凝珠上所有寡核苷酸的数目的)。(见下图1)
在液滴中,细胞破裂,磁珠上的DNA寡核苷酸捕获并标记释放的转录本;随后液滴破裂,所有细胞一起进行、逆转录、PCR扩增并通过高通量平台测序。测序得到序列与参考基因组进行比对,对应到注释的基因;再根据比对序列上的cell barcode区分来自同一个细胞序列。最后使用UMI计算每个细胞中表达的单个基因的转录本的拷贝数,从而可以生成基因表达矩阵(DGE, digital gene expression),这个矩阵就是包含了细胞barcode和基因counts的表格文件。
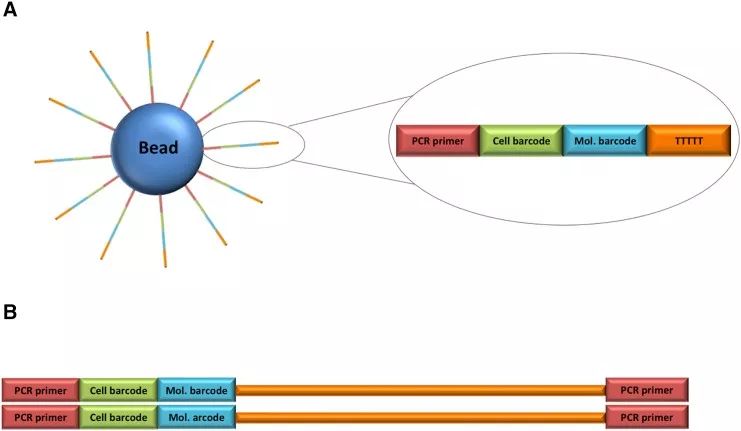
图一:Drop-Seq Bead的结构和所得序列文库
(A)DropSeq单细胞测序珠的结构。
DNA寡核苷酸的3’末端是一个poly(T)尾巴,可用于捕获细胞中的mRNA ;5’末端是用于标记细胞的cell barcode序列,一个凝珠上结合的所有寡核苷酸的cell barcode都一样;中间还有一个具有高度多样性的唯一分子标识符(unique molecular identifier,UMI),磁珠上的每个寡核苷酸的UMI都不一样。
(B)测序文库的结构。
- 红色:PCR引物,也可用作测序引物;
- 绿色和蓝色:来自珠子的细胞和分子barcode;
- 橙色:捕获的带有poly(A/T)尾巴的转录本。
从如此复杂的测序数据中得到的可靠结论取决于后续的计算分析。
大多数可用的单细胞测序算法没有图形用户界面。因此分析单细胞数据需要一些编程的基础,这样才能更好地去执行比对、聚类和可视化数据的算法。
除此之外,对目标细胞有深刻的生物学认识是很有必要的,这样才能正确地解释数据并选择合适的质控参数等。
在单细胞测序方面的生信专家需要在分析过程中对应用的算法能选择适当的阈值,并避免产生误导性的结果,才可以对其分析结果进行有意义的生物学推断。
质控指标
基于液滴的实验可以视为对单个液滴内的单个细胞进行的成千上万次的独立实验,所以必须要对数据进行质量控制(QC)去除低质量数据。而QC是通过使用不同的指标来判断并过滤掉不合格(如技术问题或细胞质量问题等导致的)的数据。
QC指标—每个细胞检测到的转录本数量或测序序列比对到参考基因组的比例
QC参数的阈值在不同分析中不一定相同,阈值的设置取决于测序的细胞或组织。
常见的QC指标是每个细胞的转录本数量或每个细胞能比对到参考基因组的测序序列的百分比。
若细胞的转录本数量低于或高于定义好的阈值,该细胞会被标记为异常细胞并从分析除去;阈值既可以由分析者自定义(例如,细胞的转录本少于20个或者超过5,000),也可以由程序自动判断(例如,转录本总数大于所有细胞平均转录本数目2倍标准差的细胞需要被移除,cells with a sum of transcripts larger than 2 SDs from the mean are removed)。
因为如果一个细胞包含大量的转录本,可能是由于doublets(即两个或两个以上的细胞悬浮在一个液滴中)造成,这种数据要从分析中除去;如果一个细胞的检测到的转录本数量很少,意味着捕获质量较差,这可能是因为细胞死亡、细胞过早破裂或者是捕获了从细胞中逸出并漂浮在细胞悬液中的随机mRNA。
也可以应用其他QC指标,例如,直接删除表达某个特定基因的所有细胞,这样可以删除不感兴趣的污染细胞,或者更复杂一些,只包含两个或更多特定基因表达达到一定比例的细胞。
在确定QC阈值时,必须考虑所分析的组织的多样性。例如,在设计实验研究血液中转移的癌细胞时,癌细胞的数量相较于正常血细胞的数量而言非常低,因此必须调整QC指标中的转录本数量(counts of transcripts)。在该组织中血细胞是优势细胞,但与活跃的癌细胞相比,它们的表达却被认为处于相对静止状态,具有相对较低的RNA量。故而如果设置阈值为删除那些转录本数量高于平均值2倍标准差的细胞,癌细胞因为转录活性比较高,就可能会被误认为是doublets,并被全部移除。(生信宝典注:相比于很多人生搬硬套Seurat示例数据中的200,2500的筛选标准,采用n倍标准差是适应性更广的方式,尤其是不关注稀有类型时。如果自己比较了解,还是需要好好看下数据分布再定标准。如果不了解,可以先松后紧,根据最后结果再回来看转录本数目异常的细胞聚类在什么地方再做评判。)
QC指标—线粒体基因的数量
另一个常见的QC参数是线粒体基因的数量。高比例的线粒体基因表达细胞处于应激状态的指标之一,因此分析中通常需要移除线粒体基因表达占比较高的细胞,因为大多数实验不研究这一类特殊状态的细胞。
但是,与转录本数量一样,此参数高度依赖于组织类型和所研究的问题。例如,由于心肌细胞的高能量需求,心脏中总mRNA的30%是线粒体,而低能量需求的组织中占比则为5%或更少。故而线粒体mRNA占30%在心肌细胞表示健康,但在淋巴细胞表示不正常。(生信宝典注:这一步筛选也不要受Seurat文档影响太深,参数都是可以改的,只要有合适的原因。最近一期的单细胞培训,这个也是讨论的重点,国内外学者济济一堂讨论这个参数选择。)
QC指标—筛选基因
根据实验的目的,也可以添加基因特异性的QC指标。在所有细胞内表达量都很低并在细胞类型之间无统计意义的基因,可以考虑设置阈值过滤掉,减少后期的计算量:设置每个细胞内的基因count阈值(例如,基因在在每个测序的细胞中,count值都小于5)或设置所有或一个细胞子集中该基因count总和的阈值(例如,所有测序细胞中的基因∑count<=300)。
虽然排除此类基因将加快计算过程,不过可能会丢失一些表达差异很小但对数据差异有贡献的基因。(生信宝典注:不排除有一些基因表达量比较低,并且较小的变化幅度就可以带来有意义的生物效果。但表达低的基因本身检测的噪音也大,比较难区分哪些是生物差异,哪些是技术差异。私以为,原文这句描述有误。)
数据标准化和归一化
在分析测序数据时,如果要对多批测序数据进行相互比较,需要消除批次效应。这些批次效应可能是由不可避免的技术差异引起的,例如将样本冷冻存放时间、反复冻融的次数、提取RNA的方法、测序深度等。
研究人员应努力保持这些实验和测序过程中的变量恒定。但是基于液滴的测序还包含数千个单独的细胞实验,因此在标准化时还必须考虑细胞特异性偏差,以便能够将一个细胞与另一个细胞进行比较。
特异性偏差是由mRNA捕获效率引起的,在所有液滴中mRNA分子没有以相同比例被磁珠捕获,这被称为“dropout events”,它也是数据稀疏的主要原因,数据稀疏将在下一段深入讨论。
此外在bulk RNA测序中,需要被标准化的多批数据几乎来自相似的生物材料(例如将血细胞与血细胞进行比较),但是在单细胞测序中,单个细胞并不属于同一类型,这就需要调整标准化的参数以保留细胞间差异,同时还要消除技术差异带来的批次效应和细胞特异性偏差。
mRNA捕获效率很低(例如,DropSeq被认为最多能捕获每个细胞10%左右的mRNA),这是液滴型单细胞测序数据的分析面临的最大挑战。由于这些“dropout events”,DGE矩阵大部分数据都会是0,这就是数据稀疏了。因此在解释数据之前,标准化和归一化至关重要。不过,这需要假设细胞在生物学上不需要严格准确(Unfortunately, this requires making assumptions about the cells that can be biologically inaccurate)。
一种可接受的标准化测序数据的方法是利用管家基因进行比较。
首先基于文献资料和对测序的生物样品的了解,选择一个管家基因用于后续标准化。假定所选的管家基因在所有细胞中均以相同的水平表达,然后对测序数据进行归一化使所选的管家基因的表达水平在所有细胞中均相等。
但是这个方法也可能不准确,因为这些持家基因在不同细胞中表达量并不总是一致的。另一个思路是基于在所有或一部分细胞中所有表达无差异的基因进行标准化。这一方法基于所有细胞或部分细胞之间表达无差异的所有基因均在所有或部分细胞中均等表达的假设进行归一化,并推断出每个细胞的归一化因子来标准化转录本的计数。
降维和可视化
PCA
对基因表达谱标准化后,应用无偏聚类的算法可以确定哪些细胞更为相似。
Principal component analysis(PCA)通常是首选的聚类算法,因为它是一种相对简单的线性降维算法,可以预测多维数据的相关性,具体的在单细胞分析中指只需要依赖高可变基因的表达谱就可以预测细胞间的相似关系。
PCA把相关的基因合并到“metagene”或主成分(PC)中。PC1解释最大的数据差异,具有最大的标准差(例如对于一个实验,细胞之间30%的差异由定义了PC1的基因解释),PC2解释了数据的第二大部分差异(例如,细胞之间20%的差异可归因于PC2中的基因,而8%则归因于PC3中的基因),然后依此类推,简单来说PC的排名就是解释数据差异贡献的顺序,其中PC1是排名最高的PC,同时也说明PC排名越低,对解释数据差异的贡献就越小。
使用排名较低的PC一般都没什么好处,因为它既增加了计算量,又几乎没有将任何信息添加到细胞间差异的展示中。因此,决定用于可视化的PC数非常重要。常用的判断方式就是绘制knee图或elbow图,如下图所示。
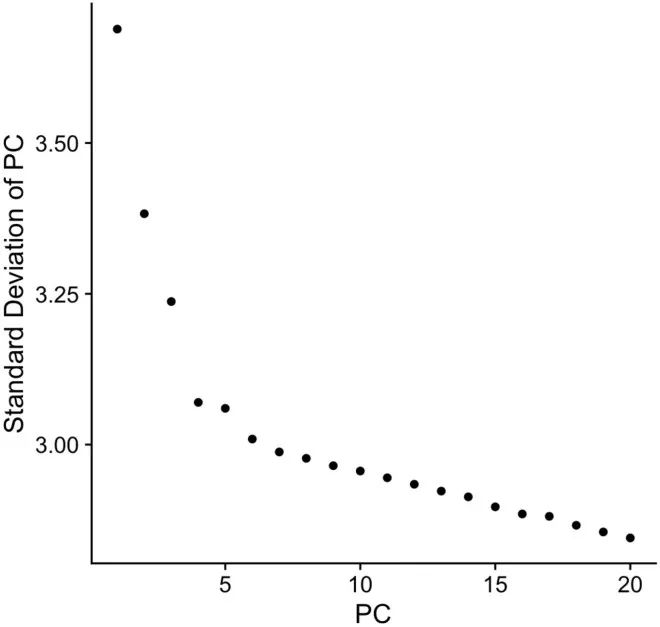
图中展示了每个主成分的标准差,代表每个PC对数据差异解释的贡献度。PC4、PC5和PC6都在拐点附近,说明推荐使用前四个、前五个和前六个主成分用于后续分析。
t-SNE
t-Distributed stochastic neighbor embedding(t-SNE)是一种常见的可视化方法。
它使用机器学习的算法来降低维数,非常适合将高维数据放到二维或三维空间中可视化展示,并且不会丢失细胞之间的相对距离的信息。
例如,如果发现用七个PC可以很好地表示细胞的多样性,就得需要七个轴或维度来展示细胞的空间分布。t-SNE能维持细胞在七维空间的关系并在二维图上展示细胞,所以在七维图上相邻的细胞在二维图上仍然相邻。同时PCA分析是线性的,t-SNE是非线性降维方法。
注意事项:有关数据生成效率和可替代的单细胞平台
在本篇综述中讨论的计算方法主要是用于基于液滴的分离方法,例如DropSeq和Chromium 10X。
不过大多数单细胞测序平台都是用特异的DNA barcode标记每个细胞的mRNA,从而得到每个细胞的基因表达信息的,所以上述介绍的类似的原理和算法也可用于其他方法的数据集。但是要注意不同平台之间始终存在着技术的区别或仪器的差异。
Valihrach等人发表了一篇综述(https://www.ncbi.nlm.nih.gov/pubmed/29534489),详细地描述了细胞分离、标记和DNA扩增的平台和方法,并且讨论了每种平台的基本原理以及不同方法的优点和缺陷。
例如,在SCI-Seq中,先使用酒精固定细胞,使其具有渗透性,再使用流式细胞仪对固定的细胞进行分选,最后将特定数量的细胞分配到多孔板的每个孔中(见表1)。
每个孔中细胞的mRNA均通过反转录结合了该孔特有的寡核苷酸,然后合并所有孔中的细胞,并以较低的密度对细胞进行另一轮荧光激活细胞分选(FACS),然后添加第二个独特的孔特异性barcode,为每个细胞创建唯一的barcode组合。此过程可以再次重复,帮助降低同一barcode的组合标记两个细胞的可能性。
这种barcode组合标记单个细胞的方法需要专门的算法来得到DGE矩阵,因为与基于液滴的方法相比,单个细胞不是由单个barcode确定,而是由barcode的特定组合确定的。值得注意的是,由于这种方法至少需要两轮细胞分选,可能会对细胞产生更大的影响并且影响基因的表达。
另一个示例是设置转录本/细胞参数的数目以排除doublets,因为每种方法都会有不同的doublets比例。
在Fluidigm C1系统中,单个细胞被隔离在特定大小的区室捕获,在隔离的中等大小的96-区室中对细胞进行显微镜检查后,doublets的比例从7%下降至3%。这个比例没有为0是因为细胞有时会在隔离室中相互堆叠,使它们看起来像单个细胞,显微镜检查就无法发现这种堆叠的细胞。
如果经显微镜检查后还有3%以上的细胞或未经检查的数据中有7%的以上的细胞的转录本的数量显著更高(例如比转录本的平均值高2倍的标准差以上),这表明可能这批细胞是由少数的有转录活性细胞和大多数的无转录活性的细胞组成,或者可能是由于doublets的比例很高,如果是在这种情况,可能需要换更小的隔离室来选择细胞了。
当前主流的单细胞分析流程是以细胞之间表达差异最大的基因为基础。这有助于发现未知细胞类型的基因marker。但是如果研究人员打算研究非常相似的细胞类型,或想从一种主要细胞类型中找到亚型,则可以在分析之前对这些细胞进行分选和增加感兴趣的细胞数量,从而提高分析精度。
即使荧光激活细胞分选(FACS)被证明对基因表达的影响很小,分选仍延长了细胞不在最佳培养条件下而在单细胞悬浮液中的时间,这可能会对细胞造成影响并可能改变mRNA和线粒体mRNA的表达。此外,使细胞通过小区室、微流控分选仪或细胞分选仪会导致应激反应和影响对应激更敏感和更容易死亡的一些细胞类型。因此,在基于液滴的单细胞测序实验中,自身比较脆弱的细胞亚型等可能很难被发现,特别是如果在单细胞分离之前对细胞进行了分选。
结论
在本篇综述中讨论了一些重要概念,这些概念在单细胞基因表达数据分析和根据细胞类型或条件来选择参数非常重要。另外还提供了一些其他类型技术的示例,使得分析可以用于基于非液滴的单细胞测序数据。
分析流程首先从原始测序文件生成包含每个细胞的基因计数DGE矩阵开始;接下来通过QC除去可能由于doublets和细胞应激等产生的错误细胞;再进行标准化和归一化解决不可比的问题(由mRNA捕获率低导致);然后基于细胞间高可变的基因进行降维和聚类;最后在二维或三维空间上展示数据中每个细胞与其他细胞的相关性。
通用算法,通常这些算法包含在易于使用的程序包中:
- Seurat,这是一个基于R的程序包,可创建与其他下游算法兼容的R对象(https://satijalab.org/seurat/);
- scran,还包括用于细胞周期分配的算法(http://bioconductor.org/packages/release/bioc/html/scran.html);
- ascend,其中包括完善的新算法,提供了灵活的分析框架(https://github.com/IMB-Computational-Genomics-Lab/ascend)。
评估以上和其他一些软件包检测到的高变基因的准确性和精确性:https://www.ncbi.nlm.nih.gov/pubmed/29481632
后续分析算法,基于实验目的,可选的更有针对性地下一步分析的算法:
- Monocle,该算法旨在分析单个细胞的分化轨迹(http://cole-trapnell-lab.github.io/monocle-release/);
- SingleSplice,用于研究单细胞群体中的可变剪接(https://github.com/jw156605/SingleSplice);
- OncoNEM,这是一种推断肿瘤细胞之间基于层次进化关系的工具(https://bitbucket.org/edith_ross/onconem/src)。
单细胞测序工具及其应用的大集合可在下列网站上找到,这些网站也会更新有最新推出的工具:
- https://github.com/seandavi/awesome-single-cell
- https://www.scrna-tools.org/
来自外部的引用