

Since version 2.3, newbler has a -cdna option for de novo transcriptome assembly. In this post, I’ll explain the principles and setting up the transcriptome assembly. The next post will discuss the output of a transcriptome assembly.
1) Principles of transcriptome assembly
As with other assembly projects, the first steps for transcriptome assembly are identical, and newbler builds a contig graph, see this post. Ideally, the reads coming from the transcript of a certain gene should result in a single contig. However, because of splice-variants (and other sequence particularities), there may be several contigs for each transcript, which themselves form a small contig graph. Splice-variants will result in reads that , relative to other reads have an insert (representing an additional exon in the transcript), thereby breaking the contig graph, see the figure.
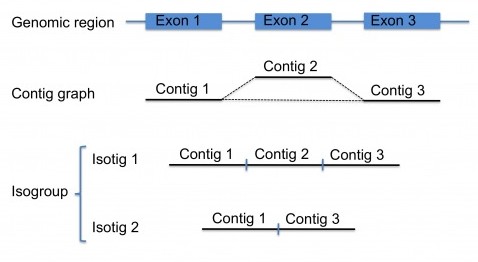
So, for transcriptome projects, there will be numerous subgraphs each potentially representing one gene. Each of these subgraphs are called an isogroup. Next, newbler will traverse the contigs in the subgraphs of each isogroup to generate transcript variants, which are called isotigs, again, see the figure. There are certain rules for this traversing step, for example, for starting the path and for ending it. Another rule, for complex graphs, is a cutoff such that no more than a maximum number of isotigs are generated per isogroup (by default set to 100 isotigs). If fully traversing the graph will result in more isotigs than this maximum, the contigs of this isogroup are reported in the output instead of the isotigs.
Ideally, these isotigs represent alternative splice-variants, and the contigs represent the exons. However, primary transcripts can result in intronic contigs, and untranslated regions (5’ UTR and 3’ UTR) will also be represented among the contigs. Furthermore, in my own experience, sequence variants between reads coming from different sister chromosomes, or from different individuals, will result in two or more almost identical contigs for a region of the transcript, resulting in an inflated number isotigs, some of which are nearly identical. A solution to this problem would be to cluster the isotigs for each individual isogroup (for example, by using CD-HIT) something I unfortunately never got to trying…
Finally, there will be three types of isogroups:
- those with two to many isotigs
- those with only one isotig consisting on one contig (no variants)
- those with only contigs and no isotigs
2) Setting up a cDNA assembly
Basically, the usual way of running newbler is used, except that the -cdna flag is added on the command line, e.g.:
runAssembly -cdna -o cdna_project1 /data/sff/*.sff
There is an additional set of parameters available for transcriptome assembly:
-ig
Isogroup Threshold: the maximum numbers of contigs in an isogroup (default 500)
-it
Isotig Threshold: the maxmimum number of isotigs in an isogroup (default 100, maximum 10000).
When the -ig and -it limits are exceeded, instead of generating isotigs, newbler will output the contigs for that particular isogroup
-icc
Isotig Contig Count Threshold: the maximum number of contigs in an isotig (default 100, maximum 200). For isotigs that exceed this maximum, no isotig is generated.
-icl
Isotig Contig Length Threshold. Isotigs containing contigs shorter than this length will not be generated (default 3 bp, minimum 3 bp).
If an isotig is skipped because of the -icc or -icl thresholds, and any of the contigs of the skipped isotig are _not_ in another isotig, then they will be added to the output.
Notes:
1) that for transcriptome assemblies, the minimum length of contigs to be reported in the 454AllContigs.fna file is set to 0 and can not be overridden using the ‘-a’ flag.
2) the manual specifically states not to use the ‘- large’ option for transcriptome assemblies, unless “the project’s computation won’t complete without using the option”. In my experience, many a transcriptome assembly would not have finished ‘before Christmas’ had I not added the -large option. So, I recommend using it, and then testing if the assembly might finish without (which in theory should result in a better assembly).
1F
正在找这方面资料呢,楼主辛苦了