

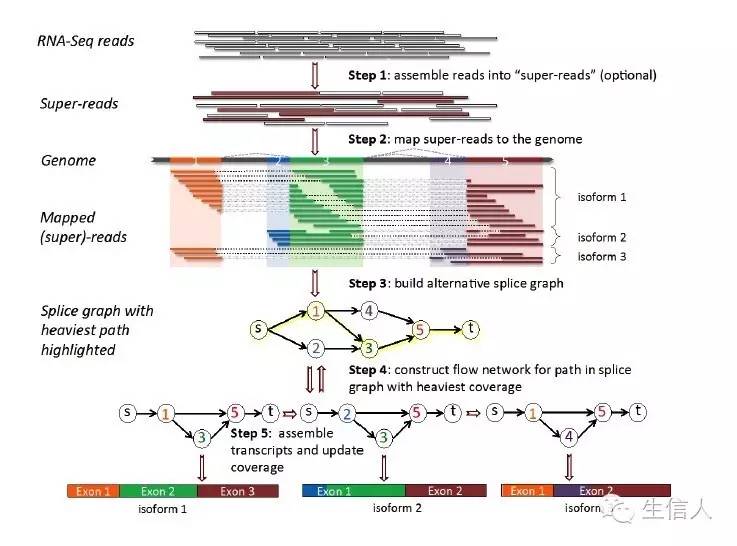
StringTie是一个新的转录组标表达定量软件,下面是组装算法示意图
摘要
转录组项目测序往往会产生2亿多得read。我们引入了StringTie软件,他利用了最优化算法(生信科班出身的应该必修课)中研究比较多的网络流算法,并且兼顾从头组装方法,来讲这些复杂的数据组装成为转录本。当对模拟数据和真实数据(人的四个组织)进行分析时,相对其他转绿本组装软件比如Cufflinks,IsoLasso,Scripture,Traph等,StringTie的结果往往更准确,更加完整,同时对于基因表水平的评估也更为准确。举例,对于人血液中90百万的read数据,它成功的组装出10990个正确的转录本,而cufflinks(排第二)只有区区7187个,相对而言有53%的提升。对于模拟数据,组装出7559相对cufflinks 6310 有20%的提升。另外在处理同一批数据的时候,StringTie相对其他软件要快的多。
研究背景
过去十年间转录组数据越来越多,并且作用也越来越重要。即使是不编码的有调控能力的ncRNA也是火得一塌糊涂。对于一个基因组而言,大概有90%的基因(多外显子的,单外显子不存在这种情况,因为只有一个外显子)有多个转录本,30%的ncRNA有可变剪切。对于人的研究,应该是比较多的,尚且不能研究透彻,对于其他基因组清晰的认识还是有很长的路要走。
近年来很对发现基因的工作也被RNA-seq改革掉了,比如原来要利用好一切统计学模型对基因的模型组装,可变剪切,编码区,其他信号位点识别,现在统统被可以利用RNA-seq数据的软件废弃掉了。话说回来,转录组数据太多,转录本的组装挑战很大。具体问题有
(1)不同的转录本read覆盖度不同
(2)可变剪切的转录本共享exon
这些问题会使组装多重异构体变的很难。表达定量同样困难重重,即使我们知道正确的基因结构。共享exon、错误比对、低表达都会影响定量效果。
尽管有很多软件研究这些方面,比如转录本组装的Trinity,定量软件RSEM,或者组装和定量一起做的软件cufflinks等,但是仍然需要付出很大的努力来得到更好的结果。
最近的一项研究表明这些软件即使能将一个转录本的所有的结构全部预测正确,但是可能仍然组装不出来。另外多转录本的表达和新的可变剪切都会影响现有软件的准确性。因此我们介绍StringTie软件,能有效的处理这些问题。
组装软件算法概述
转录组组装有两种主要的方法:有参(cufflinks,准确性较高,分为两种方法一种是read比对(Tophat2)基因组,一种是read 聚类,然后在聚类区域预测),无参(准确性较低)
StringTie是借组基因组指导的软件,同时又有从头组装的思想,StringTie输入文件不仅仅是tophat的bam比对文件,还可以输入super read比对的结果(利用MaSuRCA将read按照kmer切割然后两端延伸,组装成的super read, 简称SR)然后利用String+SR 来代表操作是利用了read 和super read 两种数据。
(1)将read比对到基因组上,一般都利用splice grape 过程如下图。
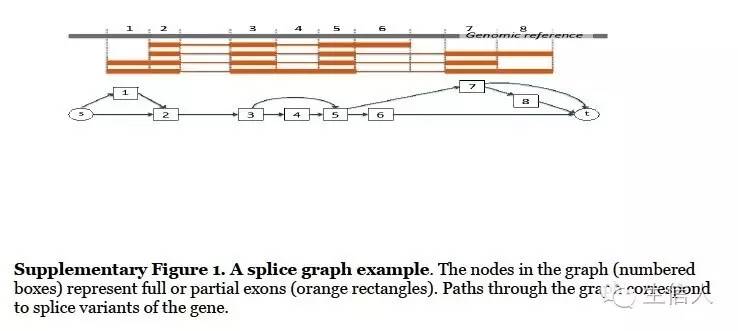
cufflinks 是典型代表,他利用吝啬法利用read来产生最少的转录本,尽管效果还行,但是异构体过多时,效果会变差。如图:
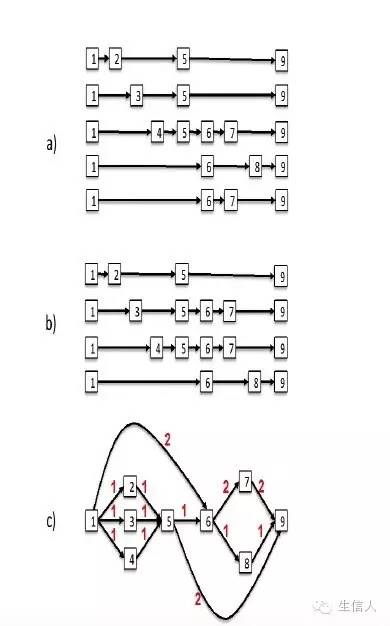
真实情况是(a),(b)。如果我们利用吝啬法去寻找,只能找到(b)结果,(a)会全部丢掉(因为read覆盖度不够。)
(2)相对于找最小集的cufflinks(定量也是分开的),StringTie组装和定量几乎并行。算法大概为首先利用聚类,然后利用read cluster 组建splice graph,然后对每一个转录本进行分别分析,利用的算法是最大网络流算法。
最大网络流在最优化算法中研究较多,但是鲜有在转录组组装和定量中使用,Traph利用到了网络流算法,但是它利用的最小网络流算法,效果不如最大网络流。
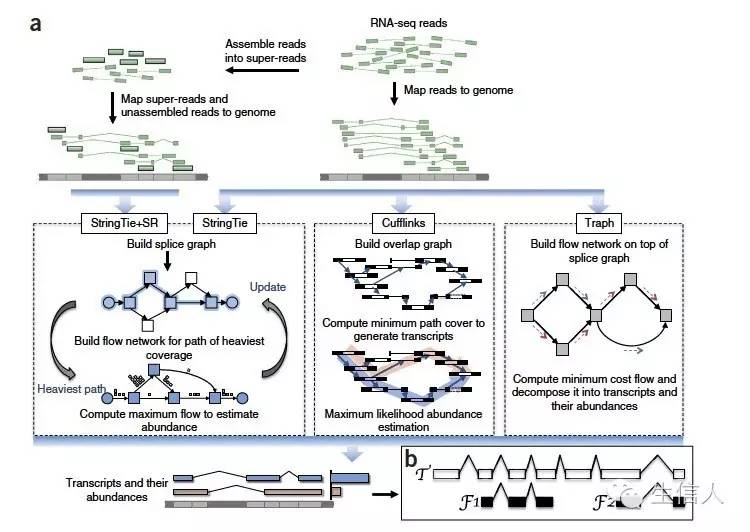
不同软件组装过程比较
数据准备
在验证软件准确性上,我们利用人的四个组织和两组模型数据对StringTie和cufflinks等其他软件预测准确性进行评估。
在谈到为什么利用模拟数据,因为相对而言模拟数据的基因结果可以预先得知,容易判断。在综合考虑模拟数据逼真度和实验设计的基础上,模拟了两套数据,一套服从现有数据的经验分布,一套服从正态分布(cufflinks设计的假设,数据服从正态分布)。
准确性标准设置
1、和真实的完全比对。
2、在起始和终止位置处相差不超过100bp(标准放宽的原因在于转录组中经常出现覆盖度的骤降,这样对于边界的识别非常困难)
3、如果本身read数据不足,只能组装出转录组的一部分,那么这些分散的部分全被组装出来就认为正确。
其中1、2考虑链特异性。
结果
模拟数据中sim-I是服从经验分布的,StringTie对基因和转录本的敏感性和阳性检测率都很高(注意不是特异性,以后分享下阳性检测率和特异性的区别)
在sim-II 中,cufflinks效果会好点,只比StringTie低一点,这也不足为奇,因为sim-II 更加适合cufflinks设计。其他软件都很差。
(a)图考虑所有的比对情况,(b)只考虑不考虑来自partial transcript的情况

在人的血液,肾脏,肺和单核细胞中对基因和转录本检测的敏感性和准确性性,如下图:StringTie 都表现的很好。
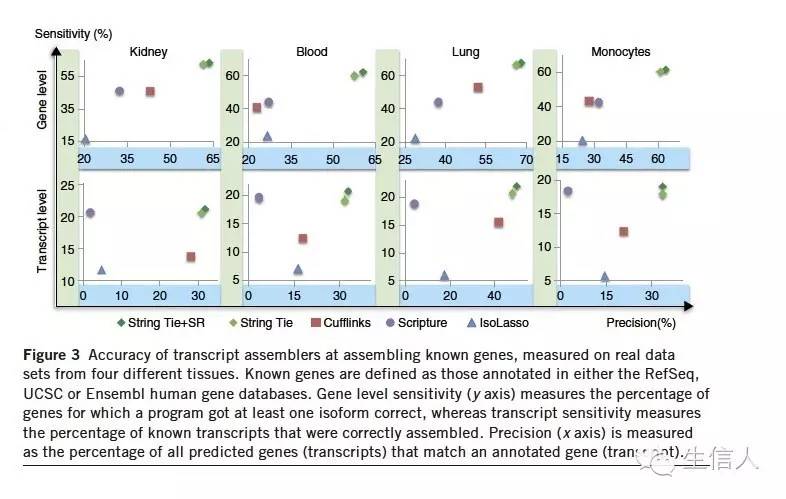
具体表格如下
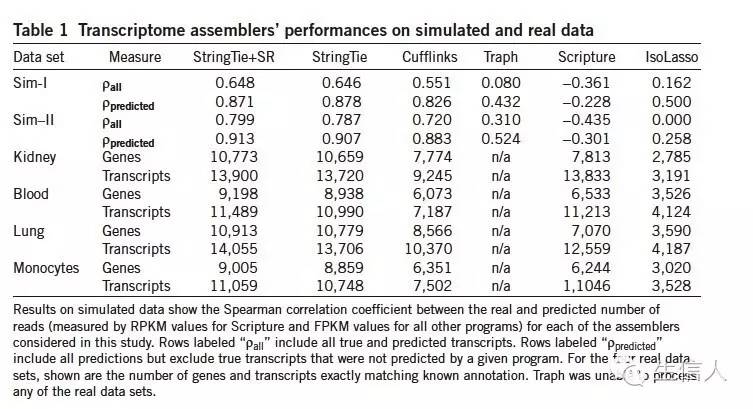
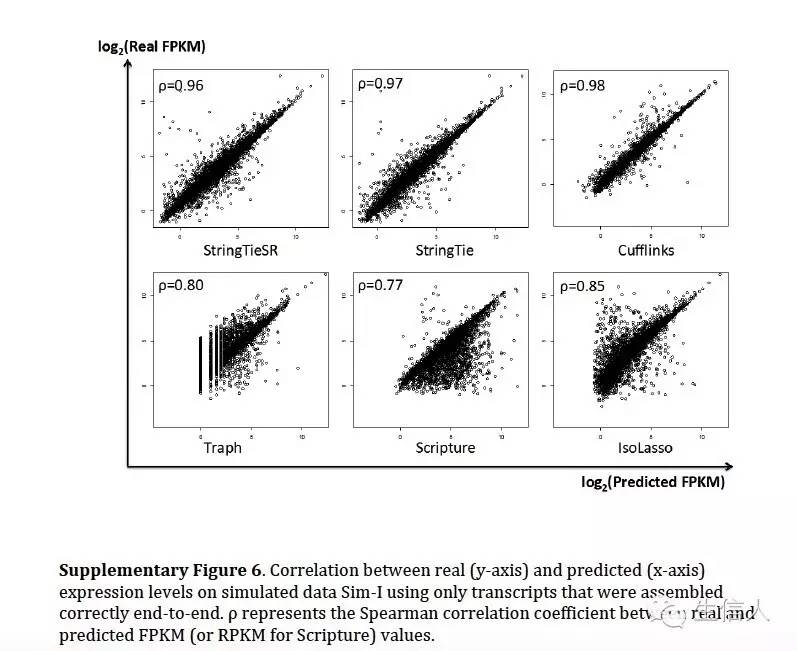
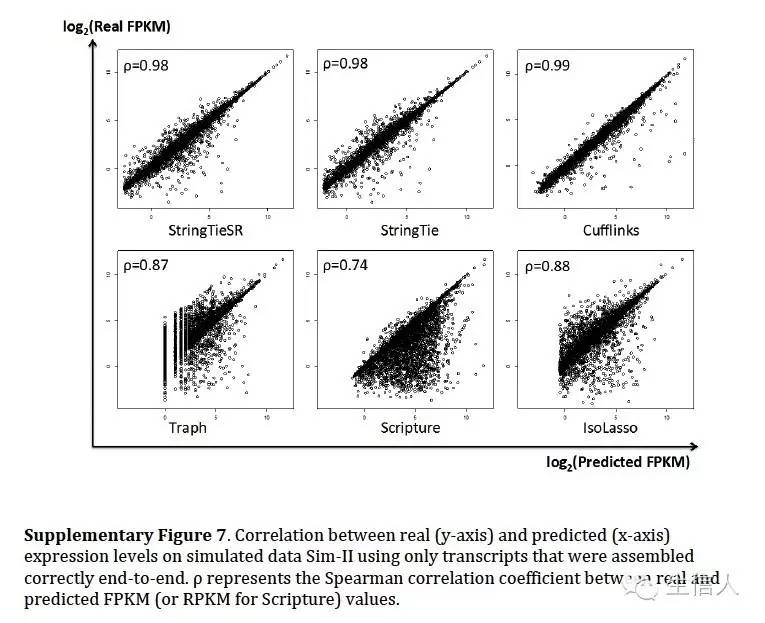
在模拟数据评估中利用了斯皮尔曼相关系数。评估真实和预测之间的相似性。
其中Pprediction代表软件预测出来的结果和真实性的相似性(参考也只是预测出来的部分),Pall代表跟所有的真实的相似性(没有预测出来的也要考虑上)
发现软件考虑到所有结果,效果很差,但是只评估预测出来的部分,效果还不错。
然后在表达定量方面,评估了真实表达值和各个软件估计表达值得情况,模拟数据上,cufflinks 相似性很高,但是StringTie 效果也可以,可以说水平旗鼓相当。
时间复杂度和空间复杂度
StringTie处理模拟数据是耗时31分钟,而cufflinks 用时81分钟,在真实数据上,耗时在35-76分钟,速度大概是其他软件的3倍左右。
在内存使用方面,处理真实数据时,大约利用了1.6~12g,cufflinks等软件利用了6.4~26.6g。
结论
StringTie相对于cufflinks来说,对于有多重异构体的物种更擅长。我们建议在大部分的RNA-seq分析中可以利用StringTie代替cufflinks,以期得到更好,更快的结果。