RAD-seq和GBS是什么关系
简化基因组的测序方法
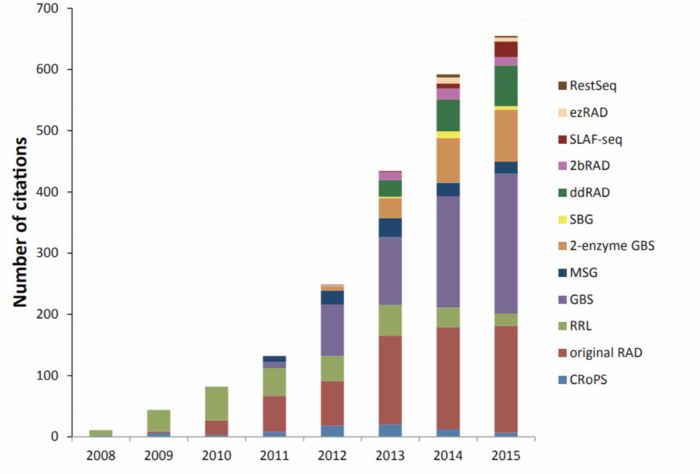
RAD-Seq(restriction site-associated DNA sequencing)最开始指的是2008年发表在PLOS ONE上“Rapid SNP discovery and genetic mapping using sequenced RAD markers”提出的方法,目前该文章的引用已经达到1200 ,现在指代的是一系列基于限制性内切酶的测序技术。同样在概念上被引申的还有GBS(genotyping-by-sequencing),只不过GBS的名字不能让你直接把它和限制性内切酶联想起来.总之,如果现在公司给你推荐GBS或RAD-seq时,可能未必和你想的一样,你需要仔细问下他们的建库手段。毕竟手段不同,你的实验设计,操作和结果都会发生变化。这是RAD-seq相关方法的历年引用情况
RAD-seq虽说方法很多,但是文库构建流程大致如下,不同方法在其中某些步骤存在差异
- 起始基因组DNA量:能否允许降解FNA
- 限制性内切酶酶解:限制酶种类,数量
- 酶切位点结合接头:接头类型
- 酶解片段大小选择:直接选择,间接选择
- 添加barcode混池:视v接头而异
- 测序类型选择:单端,双端
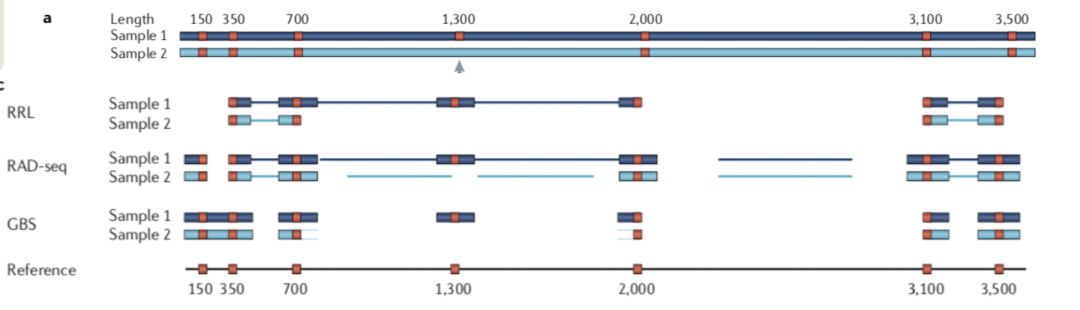
两者的差异在于,1)是现进行酶切然后随机破碎,最后仅选择存在酶切位点片段测序;2)也是酶切,但是后续直接选择合适大小的片段测序。
因此相对于1)测序的位点平均会少一点,也就会导致同一批样本后者利用率低于前者。无参考基因组更推荐前者,而不是后者。
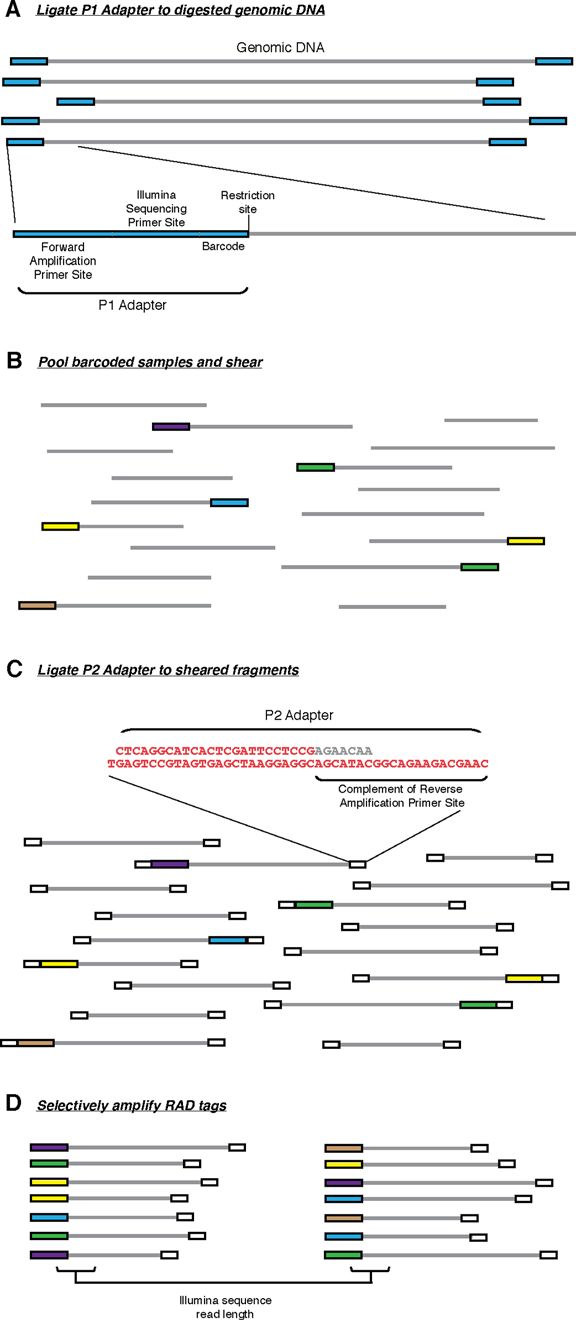
原始RAD-seqs
最先提出的RAD-seq技术流程,也就是RAD-seq的冠名技术,分为如下几步:
- 基因组DNA用限制性内切酶裂解, 然后连接到P1接头。P1接头里含有正向扩增和Illumina测序引物位点,以及4~5 bp 的核酸barcode. barcode至少大于3 bp。
- 之后接头连接的片段(adapter-ligated fragments)混池,随机打断
- DNA随后连接到P2接头,反向扩增扩展引物无法连接P2. P2是一种Y型接头,包含P2反向扩增引物位点的反向互补序列,使得不含P1接头的片段无法扩增。(Y型接头的工作原理)
- 最后仅有同时含P1和P2接头的片段能够上机测序。
Genotyping-by-Sequencing
GBS比原始的RAD-seq步骤更加简单
- 将不同样本和含不同barcode接头成对放在平板里
- 使用ApeKI限制酶进行酶解
- 使用T4连接酶,将接头连接到片段两端因酶切产生的粘末端(stcky end)
- 将含不同barcode的样本混池,随后过片段长度筛选柱,过滤尚未反应的接头
- 加入PCR引物,进行PCR扩增
这里没有直接对片段进行筛选,但是PCR扩增时优先扩增小片段
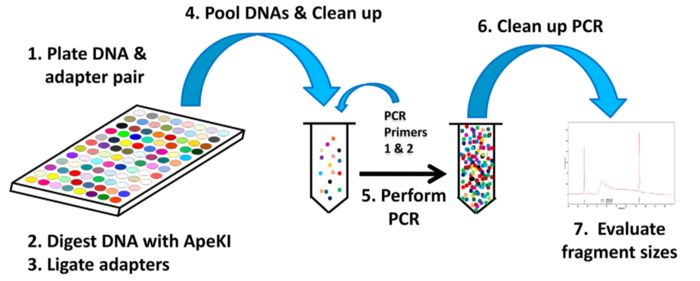
Genotyping-by-Sequencing流程
ddRAD-seq
ddRAD-seq和GBS相似,两者都不需要在加接头后进行随机打碎,GBS通过PCR扩增的方式过滤了大片段,而ddRAD-seq通过双酶切的方式,然后筛选固定长度来选择合适大小的片段
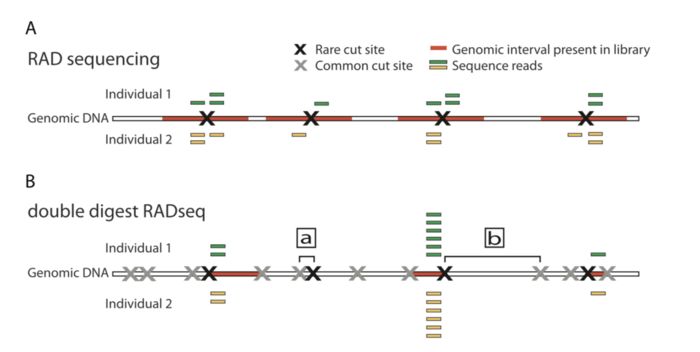
ddRAD-seq和RAD-seq的不同
常见方法的比较
其实这些RAD-seq文库制备方法可以简单的分为两类:
- 1)对单酶切位点邻近片段测序,如最初的RAD-seq
- 2)对酶切位点两翼片段测序,如Genoytping-by-Sequencing
下面是常见的物RAD-seq方法比较
| 方法 | 原始RAD | 2bRAD | GBS | ddRAD | ezRAD |
|---|---|---|---|---|---|
| 控制位点的方法 | 选择限制酶 | 选择限制酶 | 选择限制酶 | 选择限制酶和片段大小选择阈值 | 选择限制酶和片段大小选择阈值 |
| 位点数/Mb | 30~500 | 50~1000 | 5~40 | 0.3~200 | 10~800 |
| 位点长度 | 300bp 或1kb contig | 33–36 bp | < 300 bp | < 300 bp | <300 bp |
| barcode费用/样本 | 低 | 低 | 低 | 低 | 高 |
| 添加barcode难度/样本 | 中等 | 低 | 低 | 低 | 高 |
| 是否用到专利试剂盒 | 否 | 否 | 否 | 否 | 是 |
| 识别PCR重复 | 使用双端测序 | 不能 | 使用降解的barcode | 用降解的barcode | 不能 |
| 特殊的设备 | 超声破碎仪 | 无 | 无 | Pippin Prep或普通的跑胶仪 | Pippin Prep或普通的跑胶仪 |
| 是否适用复杂和大基因组 | 好 | 差 | 中等 | 好 | 好 |
| 是否适用无参考基因组 | 好 | 差 | 中等 | 中等 | 中等 |
参考文献
- RAD-seq: Rapid SNP discovery and genetic mapping using sequenced RAD markers
- GBS: A Robust, Simple Genotyping-by-Sequencing (GBS) Approach for High Diversity Species
- ddRAD-seq: Double Digest RADseq: An Inexpensive Method for De Novo SNP Discovery and Genotyping in Model and Non-Model Species
- 2011 NATURE REVIEWS | GENETICS:Genome-wide genetic marker discovery and genotyping using next-generation
- 2016 NATURE REVIEWS | GENETICS:Harnessing the power of RADseq for ecological and evolutionary genomics
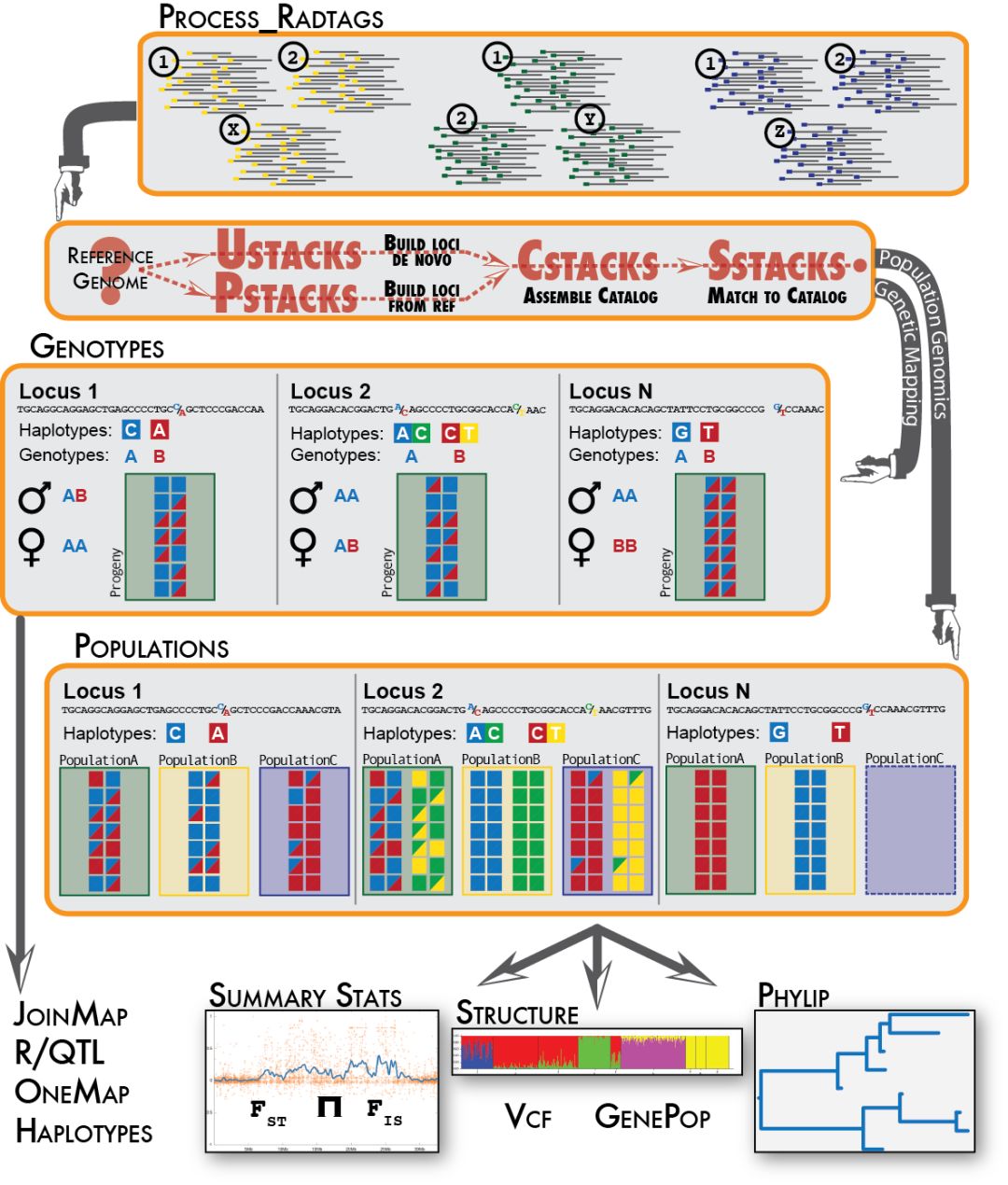
数据处理
尽管目前已经有大量物种基因组释放出来,但还是存在许多物种是没有参考基因组。使用基于酶切的二代测序技术,如RAD-seq,GBS,构建遗传图谱是研究无参考物种比较常用的方法。Stacks就是目前比较通用的分析流程,能用来构建遗传图谱,处理群体遗传学,构建进化发育树。
这篇教程主要介绍如何使用Stacks分析基于酶切的二代测序结果,比如说等RAD-seq,分析步骤为环境准备,原始数据质量评估, 多标记数据分离,序列比对(无参则需要进行contig de novo 组装),RAD位点组装和基因分型,以及后续的标记过滤和格式转换。
适用范围:
- 酶切文库类型:ddRAD, GBS, ezRAD, quad-dRAD和Rapture。 但是stacks更适用于RAD-seq,GBS推荐TASSEL。如下是“Genome-wide genetic marker discovery and genotyping using next-generation sequencing”对几种常见的建库方法的总结
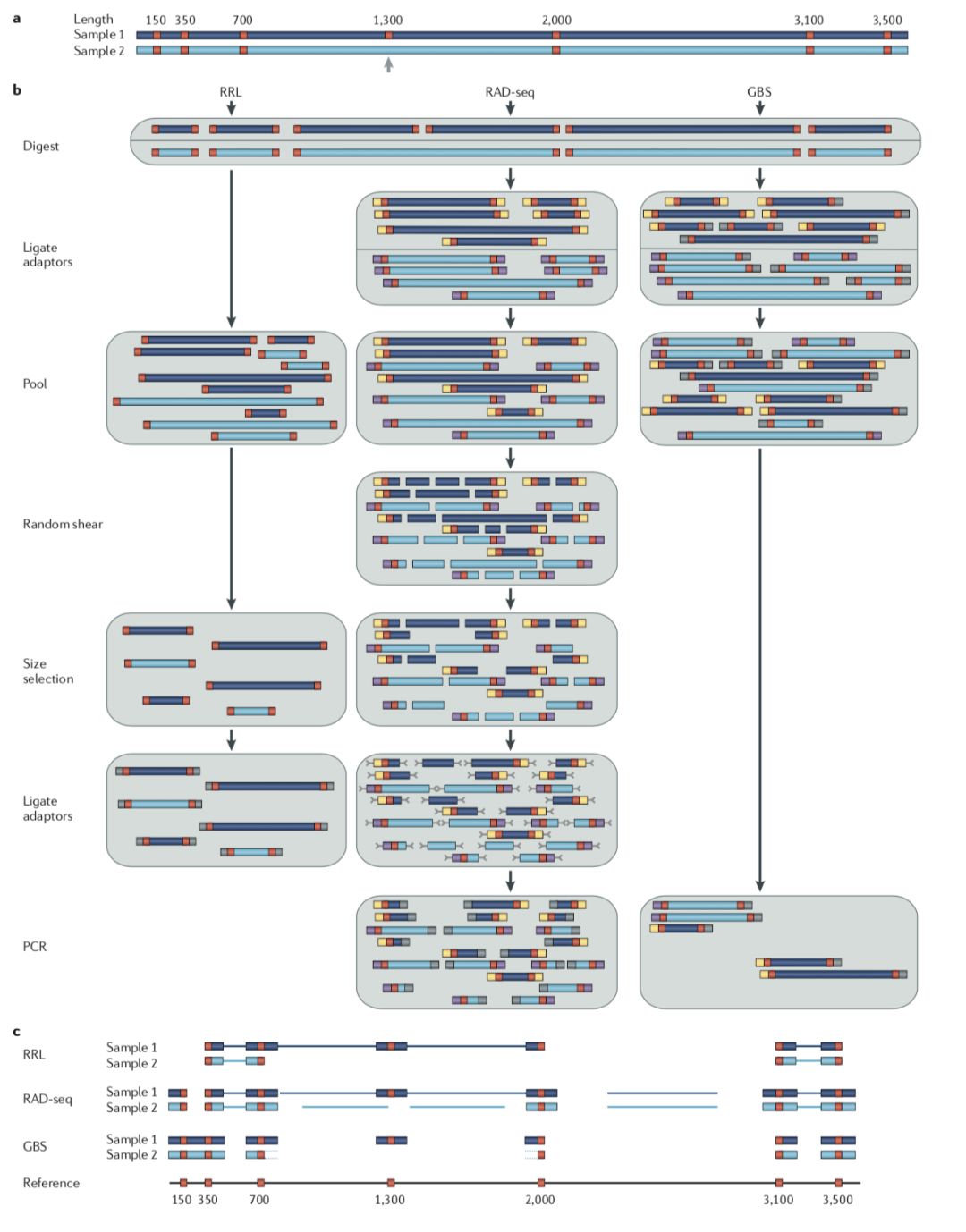
- 测序类型: 双酶切文库双端测序数据或单端数据
- 测序平台: illumina, IonTorren
局限性:
- 不能用于普通的随机文库测序
- 不能适用单酶切文库的双端测序数据(stacks 2.0可以)
- 无法用于混池测序,也不适合与多倍体,因为stacks在组装时假定物种为二倍体
- 对于深度不够,且错误率比较高的数据,它也没辙,所以建议深度在20x以上
分析者要求:掌握基本的Unix命令行,会基本集群操作,熟悉R语言编程。
硬件要求:电脑的内存在64G以上,8~16核CPU起步,准备1T以上的硬盘。
前期准备
准备分为两个部分:软件安装和数据下载。
数据准备: 数据来自于2012年发表的”The population structure and recent colonization history of Oregon threespine stickleback determined using restriction-site associated DNA-sequencing”中的三刺鱼( Gasterosteus aculeatus )数据集,一共有78个样本,来自于美国俄勒冈海岸的4个群体,两个是海水鱼(Cushman Slough’ (CS)和 ‘South Jetty’ (SJ)),两个是淡水鱼(‘Winchester Creek’ (WC) 和 ‘Pony Creek Reservoir’ (PCR))。
mkdir -p stacks-exercisecd stacks-exercise wget -q http://catchenlab.life.illinois.edu/data/rochette2017_gac_or.tar.gz tar xf http://catchenlab.life.illinois.edu/data/rochette2017_gac_or.tar.gz
这个数据大约在9G左右,因此需要很长一段时间,这段时间可以安装软件。
软件安装:需要安装BWA, SAMtools, stacks,R/ADEgenet. 好消息是这些软件都可以通过bioconda进行安装,除了R/ADEgenet推荐直接用R的install.packages("adegenet")
# 适用conda安装软件conda install -c bioconda bwa conda install -c bioconda samtools conda install -c bioconda stacks=1.47
估计此时数据依旧没有下载完,我们先创建后续需要用到的文件目录
mkdir -p stacks-exercise/{00-raw-data,01-clean-data,02-read-alignment,reference/genome,stacks/{de-novo,ref},test/{de-novo,ref},info}# 目录结构stacks-exercise/
|-- 00-raw-data # 原始数据|-- 01-clean-data # 处理后数据|-- 02-read-alignment # 比对后数据|-- info # barcode信息|-- reference # 参考基因组| |-- genome
|-- rochette2017_gac_or.tar.gz
|-- stacks # 实际运行| |-- de-novo
| |-- ref
|-- test # 测试数据集|-- de-novo
|-- ref准备输入文件(可选)
这一步并非必须,取决公司提供给你什么样的数据。对于多个样本测序,公司可能返还的是含有barcode信息原始lane数据,那么就需要从原始数据中将各个样本的数据区分开。
先将解压得到的三个lane的原始数据移动到我们的文件夹中,
cd stacks-exercise
mv rochette2017_gac_or/top/raw/{lane1,lane2,lane3} 00-raw-data接着准备两个制表符(Tab)分隔的文件,用于将barcode和样本对应,以及样本和群体一一对应。这里不需要自己写了,只需要将作者存放info里的tsv文件复制过来即可,格式如下
mv rochette2017_gac_or/top/info/*.tsv info/# barcode和样本的对应关系head -n3 info/barcodes.lane1.tsv CTCGCC sj_1819.35GACTCT sj_1819.31GAGAGA sj_1819.32# 样本和群体的对应关系head -n3 info/popmap.tsv cs_1335.01 cs cs_1335.02 cs cs_1335.03 cs
关barcode和样本的tsv中,样本的命名里不要包含空格,只能用字母,数字,”.”,“-”和”_”, 而且有意义,最好包含原来群体名的缩写和样本编号。
可视化评估测序数据
思考并记录下按照你的实验处理,你得到的read大概会是什么结构,从理论上讲,从左往右应该分别是:barcode,限制性酶切位点和后续的测序碱基。比如说案例应该现有6个碱基的barcode,SbfI限制性位点CCTGCAGG和其余的DNA序列,总计101bp
<6-nt barcode>TGCAGG
然后我们就可以用Linux自带的文本处理命令检查一下,比如说grep/zgrep,zcat head, less/zless.
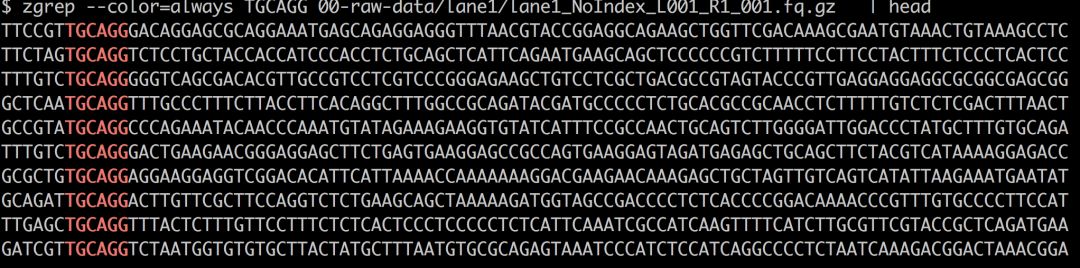
如果序列已经去掉了barcode,那么开头的就会是酶切位点。
第一步:数据预处理
这一步会用到process_radtags, 它负责对原始的数据进行处理,包括样本分离,质量控制,检查酶切位点完整性等。
# 在项目根目录下raw_dir=00-raw-data/lane1 barcodes_file=info/barcodes.lane1.tsv process_radtags -p $raw_dir -b $barcode_file \ -o 01-clean-data/ -e sbfI --inline_null \ -c -q -r &> 01-clean-data/process_radtags.lane1.oe &
解释下参数,虽然大部分已经很明了:
-p为原始数据存放文件夹
-b为barcode和样本对应关系的文件
-o为输出文件夹
-e为建库所用的限制性内切酶
--inline_null表示barcode的位置在单端的read中
-c表示数据清洗时去除表示为N的碱基
-q表示数据清理时要去除低质量碱基
-r表示要抢救下barcode和RAD-tag。
这一步需要留意自己的单端测序,还是双端测序,barcode是在read中,还是在FASTQ的header中,是否还需要去接头序列,是否是双酶切建库等。
另外这一步比较耗时,尽量脱机运行或者提交到计算节点中,不然突然断网导致运行终止就更浪费时间了。
将运行结果记录到日志文件中,方便后期检查报错。
运行结束后,在01-clean-data下会有除了process_radtags.lane1.oe外,还会有process_radtags.lane1.log,前者记录每条lane的数据保留情况,后者记录每个样本的数据保留情况。可以将后者复制到Excel表格中,用柱状图等方法直观了解
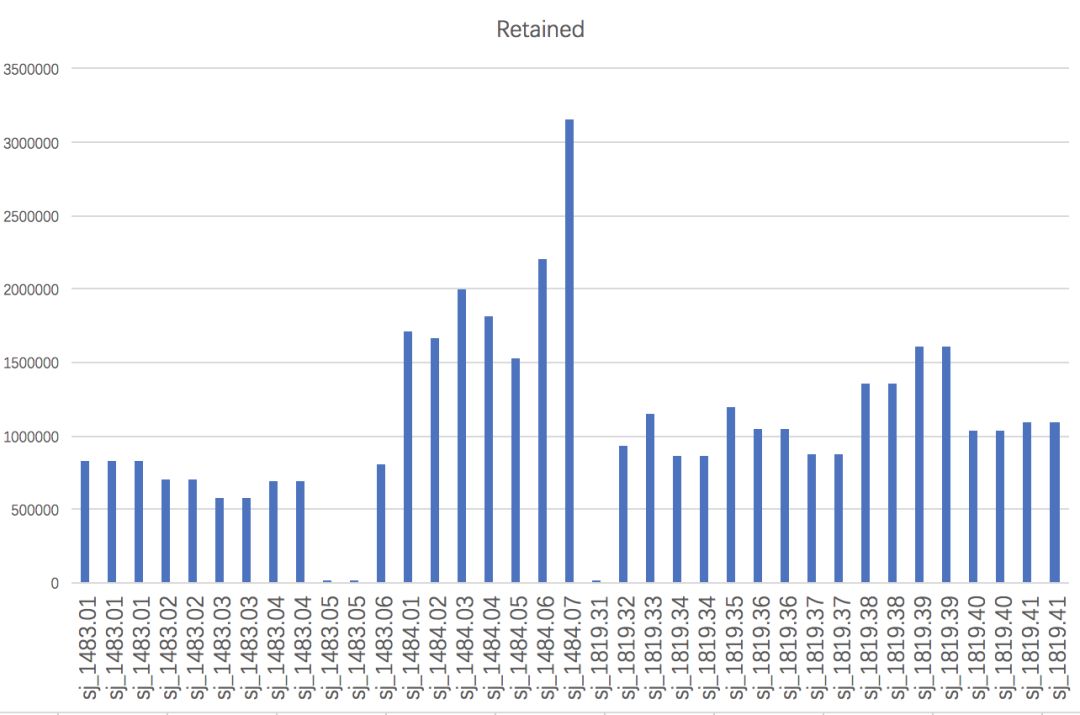
从图中可以发现,”sj_1483.05”和”sj_1819.31”几乎没有read留下来,这能是建库上导致的问题,我们需要将其从“info/popmap.tsv”中删掉或者用“#”注释掉(推荐后者)。
在数据预处理这一步,stacks还提供process_shortreads,clone_filter, kmer_filter用于处理cDNA文库和随机切割的DNA文库,如果是RAD-seq不需要用到。
如果是双端测序,stacks1.47只能通过cat合并两个数据,而不能有效的利用双端测序提供的fragment信息。stacks似乎可以,我之后尝试。
后续就要区分为有参和无参两条路了。
参考资料
- Deriving genotypes from RAD-seq short-read data using Stacks
- Genome-wide genetic marker discovery and genotyping using next-generation sequencing
第二步:序列比对
这一步之后,分析流程就要根据是否有参考基因组分别进行分析。无参考基因组需要先有一步的 de novo 组装,产生能用于比对的contig。有参考基因组则需要考虑基因组的质量,如果质量太差,则需要进一步以无参分析作为补充。
参考基因组主要用于区分出假阳性的SNP,将snp与附近其他共线性的snp比较来找出离异值,这些离异值大多是因为建库过程所引入的误差,如PCR的链偏好性扩增。
无论是何者,我们一开始都只能用其中的部分数据进行参数测试,根据不同的参数结果作为反馈,进行调优,这一步根据你的运气和经验,还有你的算力,时间不定。毕竟超算一天,普算一年。
有参考基因组
三刺鱼是可从ensemblgenomic上搜索到到参考基因组信息
- http://asia.ensembl.org/Gasterosteus_aculeatus/Info/Index
但是质量非常一般,仅仅是contig程度,只能说是凑合使用了。
建立索引数据库
stacks不直接参与比对,而是处理不同比对软件得到的BAM文件,因此你可以根据自己的喜好选择比较工具。目前,基因组比对工具都是首选BWA-mem,所以这里建立bwa的索引
# 位于项目根目录下 cd reference/genome wget -q ftp://ftp.ensembl.org/pub/release-91/fasta/gasterosteus_aculeatus/dna/Gasterosteus_aculeatus.BROADS1.dna.toplevel.fa.gz gzip -d Gasterosteus_aculeatus.BROADS1.dna.toplevel.fa.gzcd .. mkdir -p index/bwa/ genome_fa=genome/Gasterosteus_aculeatus.BROADS1.dna.toplevel.fa bwa index -p index/bwa/gac $genome_fa &> index/bwa/bwa_index.oe # 结果如下 |-- genome | |-- Gasterosteus_aculeatus.BROADS1.dna.toplevel.fa |-- index |-- bwa |-- bwa_index.oe |-- gac.amb |-- gac.ann |-- gac.bwt |-- gac.pac |-- gac.sa
小样本参数调优
这一步是为了调整比对工具处理序列相似性的参数,保证有绝大多数的read都能回帖到参考基因组上,因此参数不能太严格,能容忍遗传变异和测序误差,也不能太宽松,要区分旁系同源位点。对于BWA-MEM而言,几个和打分相关的参数值得注意:
-B: 不匹配的惩罚, 影响错配数,默认是4-O: 缺失和插入的gap打开的惩罚,影响InDel的数目,默认是[6,6]-E: gap延伸的惩罚,长度k的gap惩罚为’{-O} {-E}*k’, 默认是[1,1]-L: soft clip的惩罚,也就是read两端直接切掉碱基来保证匹配,默认是[5,5]
对于参考基因组质量比较高,且研究物种和参考基因组比较近,那么参数调整没有太大必要性。如果质量不要,或者所研究物种和参考基因组有点距离,那么就需要注意不同参数对结果的影响,必要时使用IGV人工检查。
让我们先以默认参数开始,处理其中一个样本
# 位于项目根目录下# 在测试文件下按照参数创建文件夹
mkdir -p test/ref/{alignment-bwa,stacks-bwa}
## bwa-mem比对
sample=cs_1335.01fq_file=01-clean-data/$sample.fq.gz
bam_file=test/ref/alignment-bwa/${sample}_default.bam
bwa_index=reference/index/bwa/gac
bwa mem -M $bwa_index $fq_file | samtools view -b > $bam_file &这里的bwa mem使用了-M参数,通常的解释这个参数是为了和后续的Picard标记重复和GATK找变异兼容。
进一步的解释,不用
-M,split read会被标记为SUPPLEMENTARY, 使用该选项则是标记为SECONDARY(次要),即不是PRIMARY(主要),既然不是主要比对,所以就被一些工具忽略掉。如果是SUPPLEMENTARY就有可能在标记重复时被考虑进去。其中split read是嵌合比对的一部分,具体概念见SAM格式解释。
对于比对结果,可以用samtools stats和samtools flagstat查看一下质量
samtools flagstat test/ref/alignment-bwa-default/cs_1335.01_default.bam #1310139 0 in total (QC-passed reads QC-failed reads) #7972 0 secondary #0 0 supplementary #0 0 duplicates#1271894 0 mapped (97.08% : N/A)
97.08%的比对率应该是很不错了,不过可以尝试降低下错配和gap的惩罚,看看效果
# 位于项目根目录下
sample=cs_1335.01fq_file=01-clean-data/$sample.fq.gz
bam_file=test/ref/alignment-bwa/${sample}_B3_O55.bam
bwa_index=reference/index/bwa/gac
bwa mem -M -B 3 -O 5,5 $bwa_index $fq_file | samtools view -b > $bam_file &
samtools flagstat test/ref/alignment-bwa-default/cs_1335.01_default.bam
#1309830 0 in total (QC-passed reads QC-failed reads)
#7663 0 secondary
#0 0 supplementary
#0 0 duplicates#1272297 0 mapped (97.13% : N/A)也就提高了0.05%,所以就用默认参数好了。通过IGV可视化,可以了解简化基因组的read分布是比较稀疏,10k中可能就只有2个。
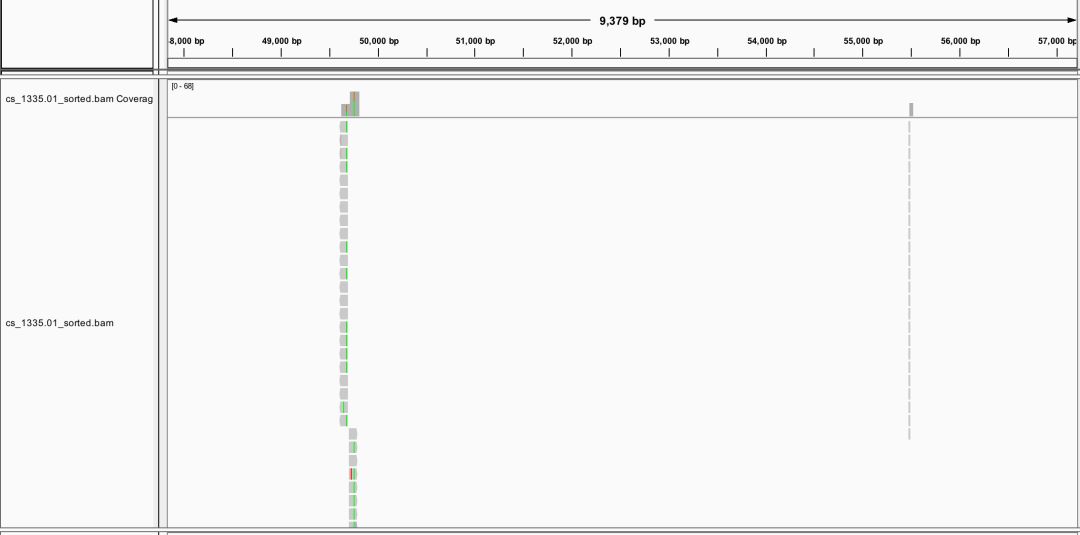
得到的BAM文件使用pstack中找snp,
# 位于项目根目录下
sample=cs_1335.01
sample_index=1
bam_file=test/ref/alignment-bwa/${sample}_B3_O55.bam
log_file=test/ref/stacks-bwa/$sample.pstacks.oe
pstacks -t bam -f $bam_file -i $sample_index -o test/ref/stacks-bwa/ &> $log_file这里的参数也很简单,-t用来确定输入文件的格式,-f是输入文件,-i对样本编序,-o指定输出文件夹。除了以上几个参数外,还可用-p指定线程数,-m来制定最低的覆盖度,默认是3.还可以用--model_type [type]制定模型。
最后得到$sample.tags.tsv.gz, $sample.models.tsv.gz, $sample.snps.tsv.gz, 和 $sample.alleles.tsv.gz共4个文件,以及一个日志文件。参数评估的主要看日志文件里的几个指标:
- 实际使用的alignment数
- 因soft-clipping剔除的alignment数,过高的话要对比对参数进行调整
- 每个位点的平均覆盖度,过低会影响snp的准确性。
这里仅仅用了一个样本做测试,实际上要用10个以上样本做测试,看平均表现,
全数据集处理
在使用小样本调试完参数,这部分参数就可以应用所有的样本。除了比对和使用pstacks外,还需要用到 cstacks根据位置信息进一步合并成包含所有位点信息的目录文件,之后用 sstacks从 cstacks创建的目录文件搜索每个样本的位点信息。代码为
cstacks -p 10 --aligned -P 03-alignment-stacks/ -M info/popmap.tsv # 以其中一个样本为例 sample=cs_1335.01 log_file=$sample.sstacks.oe sstacks --aligned -c 03-alignment-stacks/batch_1 -s 03-alignment-stacks/$sample -o 03-alignment-stacks/ &> 03-alignment-stacks/$log_file
你可以写一个shell脚本处理,不过我现在偏好用snakemake写流程,如下:
INDEX = "reference/index/bwa/gac"
SAMPLES, = glob_wildcards("01-raw-data/{sample}.fq.gz")
INDEX_DICT = {value: key for key, value in dict(enumerate(SAMPLES, start=1))}
FQ_FILES = expand("01-clean-data/{sample}.fq.gz", sample=SAMPLES)
BAM_FILES = expand("02-read-alignment/ref-based/{sample}.bam", sample=SAMPLES)
SNP_TSVS = expand("03-alignment-stacks/{sample}.snp.tsv.gz", sample=SAMPLES)
CATALOG = "03-alignment-stacks/batch_1.catalog.snps.tsv.gz"
MATCHES = expand("03-alignment-stacks/{sample}.matches.tsv.gz", sample=SAMPLES)
rule all:
input:
BAM_FILES,
rule bwa_mem:
input: "01-clean-data/{sample}.fq.gz"
params:
index = INDEX,
mismatch = "3",
gap = "5,5"
threads: 8
output: "02-read-alignment/ref-based/{sample}.bam"
shell:"""
mkdir -p 02-read-alignment
bwa mem -t {threads} -M -B {params.mismatch} -O {params.gap} {params.index} {input} \
| samtools view -b > {output}
"""
rule pstacks:
input: "02-read-alignment/ref-based/{sample}.bam"
params:
index = lambda wildcards: INDEX_DICT.get(wildcards.sample),
outdir = "03-alignment-stacks/"
threads: 8
output:
"03-alignment-stacks/{sample}.snps.tsv.gz",
"03-alignment-stacks/{sample}.tags.tsv.gz",
"03-alignment-stacks/{sample}.models.tsv.gz",
"03-alignment-stacks/{sample}.alleles.tsv.gz"
log: "03-alignment-stacks/{sample}.pstacks.oe"
shell:"""
mkdir -p 03-alignment-stacks
pstacks -p {threads} -t bam -f {input} -i {params.index} -o {params.outdir} &> {log}
"""
# A catalog can be built from any set of samples processed by the ustacks or pstacks programs
rule cstacks:
input: "info/popmap.tsv",expand("03-alignment-stacks/{sample}.snps.tsv.gz", sample=SAMPLES)
threads: 10
output:
"03-alignment-stacks/batch_1.catalog.alleles.tsv.gz",
"03-alignment-stacks/batch_1.catalog.snps.tsv.gz",
"03-alignment-stacks/batch_1.catalog.tags.tsv.gz"
shell:
"cstacks -p {threads} --aligned -P 03-alignment-stacks/ -M {input[0]}"
# Sets of stacks, i.e. putative loci, constructed by the ustacks or pstacks programs
# can be searched against a catalog produced by cstacks.
rule sstacks:
input:
"03-alignment-stacks/{sample}.snps.tsv.gz",
"03-alignment-stacks/{sample}.tags.tsv.gz",
"03-alignment-stacks/{sample}.models.tsv.gz",
"03-alignment-stacks/{sample}.alleles.tsv.gz"
params:
catalog = "03-alignment-stacks/batch_1",
sample = lambda wildcards: "03-alignment-stacks/" wildcards.sample
output:
"03-alignment-stacks/{sample}.matches.tsv.gz"
log: "03-alignment-stacks/{sample}.sstacks.oe"
shell:"""
sstacks --aligned -c {params.catalog} -s {params.sample} -o 03-alignment-stacks &> {log}
"""将以上代码保存为Snakemake,没有集群服务器,就直接用 snakemake运行吧。因为我能在集群服务器上提交任务,所以用如下代码:
snakemake --cluster "qsub -V -cwd" -j 20 --local-cores 10 &
无参考基因组分析
基于参考基因组的分析不能体现出RAD-seq的优势,RAD-seq的优势体现在没有参考基因组时他能够提供大量可靠的分子标记,从而构建出遗传图谱,既可以用于基因定位,也可以辅助组装。
和全基因组随机文库不同,RAD-seq是用限制性内切酶对基因组的特定位置进行切割。这样的优点在于降低了 de novo 组装的压力,原本是根据overlap(重叠)来延伸150bp左右短读序列,形成较大的contig,而现在只是将相似性的序列堆叠(stack)起来。这样会产生两种分子标记:1)由于变异导致的酶切位点出现有或无的差异;2)同一个酶切位点150bp附近存在snp。
参数调优
这一步使用的核心工具是 ustacks和 cstacks,前者根据序列相似性找出变异,后者将变异汇总,同样需要使用小样本调整三个参数 -M, -n, -m。 ustacks的 M 控制两个不同样本等位基因(allele)之间的错配数,m 控制最少需要几个相同的碱基来形成一个堆叠(stack).最后一个比较复杂的参数是能否允许gap(--gap)。而 cstacks的 n 和 ustacks的 M等价。
因此,我们可以尝试在保证 m=3的前提,让 M=n从1到9递增,直到找到能让80%的样本都有多态性RAD位点,简称r80. Stacks提供了 denovo_map.pl来完成这部分工作,下面开始代码部分。
首先是从 info/popmap.tsv中挑选10多个样本用于参数调试。
cat popmap_sample.tsv cs_1335.01 cs cs_1335.02 cs cs_1335.19 cs pcr_1193.00 pcr pcr_1193.01 pcr pcr_1193.02 pcr pcr_1213.02 pcr sj_1483.01 sj sj_1483.02 sj sj_1483.03 sj wc_1218.04 wc wc_1218.05 wc wc_1218.06 wc wc_1219.01 wc
然后为每个参数都准备一个文件夹
mkdir -p test/de-novo/stacks_M{1..9}然后先 测试 第一个样本
# 项目根目录
M=1
popmap=info/popmap_sample.tsv
reads_dir=01-clean-data
out_dir=test/de-novo/stacks_M${M}
log_file=$out_dir/denovo_map.oe
threads=8
denovo_map.pl -T ${threads} --samples ${reads_dir} -O ${popmap} -o ${out_dir} -M $M -n $M -m 3 -b 1 -S &> ${log_file} &代码运行需要一段比较长的时间,这个时候可以学习一下参数: -T 表示线程数, --samples表示样本数据所在文件夹,-O提供需要分析的样本名, -o是输出文件夹,之后就是-M, -n, -m这三个需要调整的参数, -b表示批处理的标识符, -S关闭SQL数据库输出。同时还有遗传图谱和群体分析相关参数,-p表示为亲本,-r表示为后代,-s表明群体各个样本。
为了确保下一步能顺利进行,还需要对oe结尾的日志文件的信息进行检查,确保没有出错
grep -iE "(err|e:|warn|w:|fail|abort)" test/de-novo/stacks_M1/denovo_map.oe
以及以log结尾的日志中每个样本的平均覆盖度,低于10x的覆盖度无法保证snp的信息的准确性
之后对每个参数得到的原始数据,要用 populations过滤出80%以上都有的snp位点,即r80位点
# 项目根目录
for M in `seq 1 9`
do
popmap=info/popmap_sample.tsv
stacks_dir=test/de-novo/stacks_M${M}
out_dir=$stacks_dir/populations_r80
mkdir -p ${out_dir}
log_file=$out_dir/populations.oe
populations -P $stacks_dir -O $out_dir -r 0.80 &> $log_file &
done确定参数
经过漫长的等待后,所有参数的结果都已经保存到特定文件下,最后就需要从之确定合适的参数,有两个指标:
- 多态性位点总数和r80位点数
- 短读堆叠区的snp平均数
尽管这些信息都已经保存在了相应的文本 test/de-novo/stacks_M[1-9]/populations_r80/batch_1.populations.log中,但是通过文字信息无法直观的了解总体情况,因此更好的方法是用R处理输出绘图结果。
第一步:提取每个参数输出文件中的log文件中SNPs-per-locus distribution(每个位点座位SNP分布图)信息.新建一个shell脚本,命名为,log_extractor.sh, 添加如下内容
#!/bin/bash
# Extract the SNPs-per-locus distributions (they are reported in the log of populations).
# ----------
echo "Tallying the numbers..."
full_table=n_snps_per_locus.tsv
header=\'#par_set\tM\tn\tm\tn_snps\tn_loci\'
for M in 1 2 3 4 5 6 7 8 9 ;do
n=$M
m=3
# Extract the numbers for this parameter combination.
log_file=test/de-novo/stacks_M${M}/populations_r80/batch_1.populations.log
sed -n \'/^#n_snps\tn_loci/,/^[^0-9]/ p\' $log_file | grep -E \'^[0-9]\' > $log_file.snps_per_loc
# Cat the content of this file, prefixing each line with information on this
# parameter combination.
line_prefix="M$M-n$n-m$m\t$M\t$n\t$m\t"
cat $log_file.snps_per_loc | sed -r "s/^/$line_prefix/"
done | sed "1i $header" > $full_table运行后得到 n_snps_per_locus.tsv用于后续作图。会用到两个R脚本 plot_n_loci.R和 plot_n_snps_per_locus.R,代码见最后,结果图如下
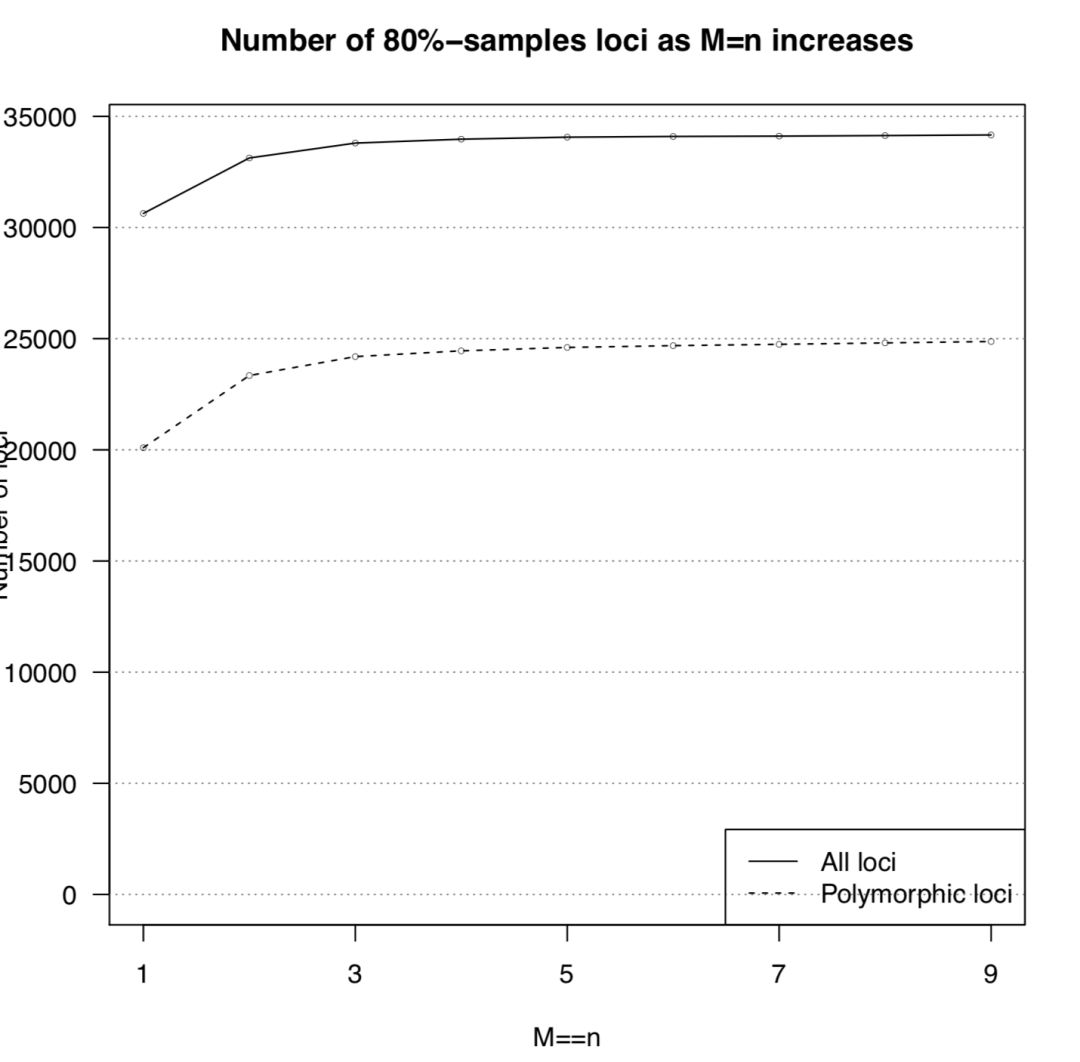
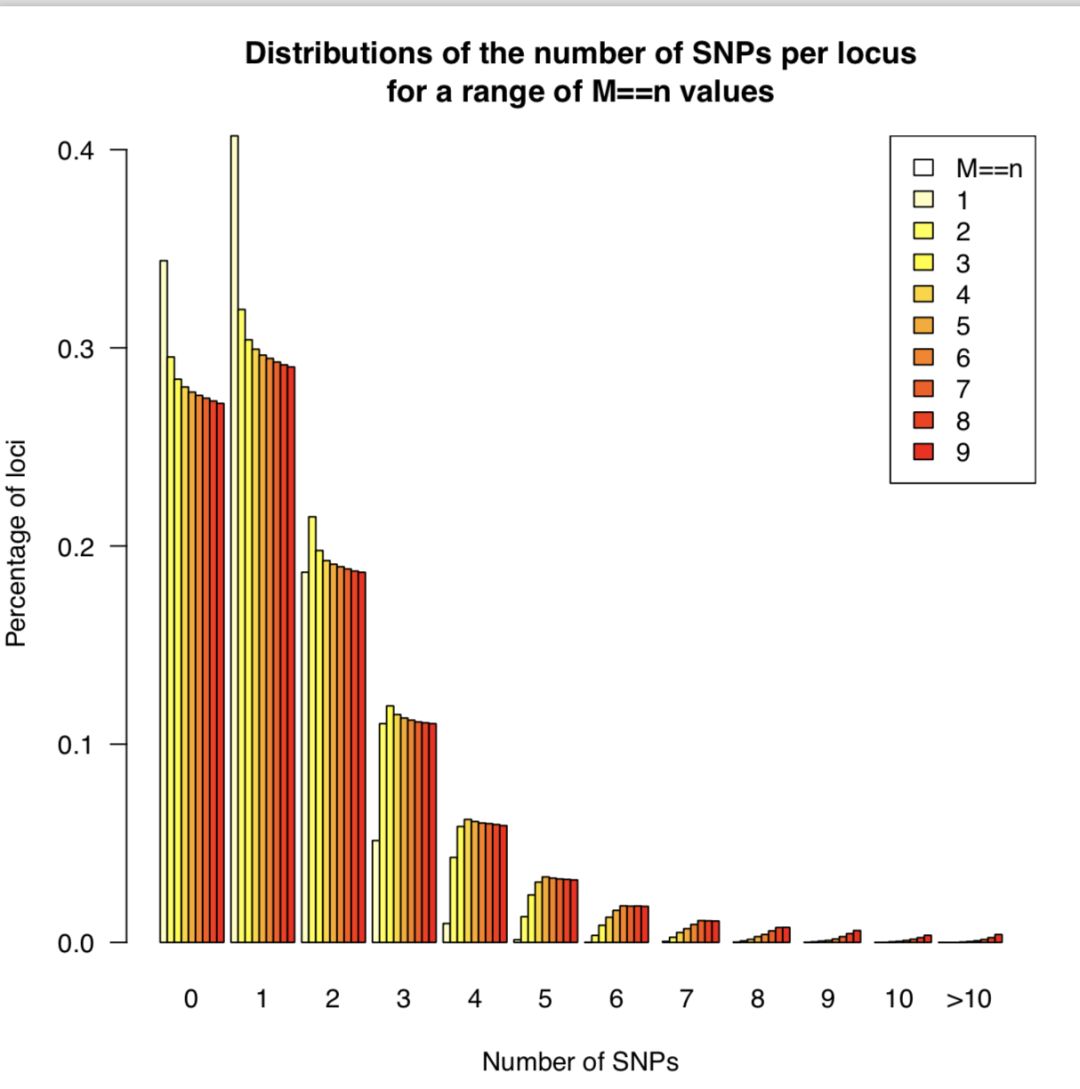
从上图可以发现M=4以后,折线就趋于稳定,并且每个座位上的SNP分布趋于稳定,因此选择M=4作为全样本数据集的运行参数。
全数据集 de novo 分析
和之前基于参考基因组分析的代码类型,只不过将序列比对这一块换成了 ustacks。尽管前面用来确定参数的脚本 denovo_map.pl也能用来批量处理,但它不适合用于大群体分析。 ustacks得到的结果仅能选择40~200个 覆盖度高 且 遗传多样性上有代表性的样本。 使用所有样本会带来计算上的压力,低频的等位基因位点也并非研究重点,并且会提高假阳性。综上,选择覆盖度比较高的40个样本就行了。
总体部分已经通过这三天讲完,明后天更新SNP过滤部分
Snakefile:
SAMPLES, = glob_wildcards("01-clean-data/{sample}.fq.gz")
INDEX_DICT = {value: key for key, value in dict(enumerate(SAMPLES, start=1)).items()}
FQ_FILES = expand("01-clean-data/{sample}.fq.gz", sample=SAMPLES)
# de novo
MISMATCH = 4 # mismatch number of ustacks
DE_NOVO_LOCI = expand("03-stacks-analysis/de-novo/{sample}.snps.tsv.gz", sample=SAMPLES)
DE_NOVO_CATA = "03-stacks-analysis/de-novo/batch_1.catalog.snps.tsv.gz"
DE_NOVO_MATS = expand("03-stacks-analysis/de-nono/{sample}.matches.tsv.gz", sample=SAMPLES)
# ref-based
INDEX = "reference/index/bwa/gac"
BAM_FILES = expand("02-read-alignment/ref-based/{sample}.bam", sample=SAMPLES)
REF_BASED_LOCI = expand("03-stacks-analysis/ref-based/{sample}.snps.tsv.gz", sample=SAMPLES)
REF_BASED_CATA = "03-stacks-analysis/ref-based/batch_1.catalog.snps.tsv.gz"
REF_BASED_MATS = expand("03-stacks-analysis/ref-based/{sample}.matches.tsv.gz", sample=SAMPLES)
rule all:
input: rules.de_novo.input, rules.ref_based.input
rule de_novo:
input: DE_NOVO_LOCI, DE_NOVO_CATA,DE_NOVO_MATS
rule ref_based:
input: REF_BASED_LOCI, REF_BASED_CATA, REF_BASED_MATS
# de novo data analysis
## The unique stacks program will take as input a set of short-read sequences
## and align them into exactly-matching stacks (or putative alleles).
rule ustacks:
input: "01-clean-data/{sample}.fq.gz"
threads: 8
params:
mismatch = MISMATCH,
outdir = "03-stacks-analysis/de-novo",
index = lambda wildcards: INDEX_DICT.get(wildcards.sample)
output:
"03-stacks-analysis/de-novo/{sample}.snps.tsv.gz",
"03-stacks-analysis/de-novo/{sample}.tags.tsv.gz",
"03-stacks-analysis/de-novo/{sample}.models.tsv.gz",
"03-stacks-analysis/de-novo/{sample}.alleles.tsv.gz",
log: "03-stacks-analysis/de-novo/{sample}.ustacks.oe"
shell:"""
mkdir -p {params.outdir}
ustacks -p {threads} -M {params.mismatch} -m 3 \
-f {input} -i {params.index} -o {params.outdir} &> {log}
"""
## choose sample for catalog building
rule choose_representative_samples:
input: expand("03-stacks-analysis/de-novo/{sample}.ustacks.oe", sample=SAMPLES)
output:
"03-stacks-analysis/de-novo/per_sample_mean_coverage.tsv",
"info/popmap.catalog.tsv"
shell:"""
for sample in {input};do
name=${{sample##*/}}
name=${{name%%.*}}
sed -n "/Mean/p" ${{sample}} |\
sed "s/.*Mean: \(.*\); Std.*/\\1/g" |\
paste - - - |\
sed "s/.*/${{name}}\\t&"
done | sort -k2,2nr > {output[0]}
head -n 50 {output[0]} | tail -n 40 | cut -f 1 | sed \'s/\([0-9a-zA-Z]*\)_.*/&\t\1/\' > {output[1]}
"""
rule de_novo_cstacks:
input:
"info/popmap.catalog.tsv",
expand("03-stacks-analysis/de-novo/{sample}.snps.tsv.gz", sample=SAMPLES)
params:
stacks_path = "03-stacks-analysis/de-novo/",
mismatch = MISMATCH
threads: 10
log:"03-stacks-analysis/de-novo/de_novo_cstacks.oe"
output:
"03-stacks-analysis/de-novo/batch_1.catalog.alleles.tsv.gz",
"03-stacks-analysis/de-novo/batch_1.catalog.snps.tsv.gz",
"03-stacks-analysis/de-novo/batch_1.catalog.tags.tsv.gz"
shell:"""
cstacks -p {threads} -P {params.stacks_path} -M {input[0]} \
-n {params.mismatch} &> {log}
"""
rule de-novo_sstacks:
input:
"03-stacks-analysis/de-novo/{sample}.snps.tsv.gz",
"03-stacks-analysis/de-novo/{sample}.tags.tsv.gz",
"03-stacks-analysis/de-novo/{sample}.models.tsv.gz",
"03-stacks-analysis/de-novo/{sample}.alleles.tsv.gz"
params:
catalog = "03-stacks-analysis/de-novo/batch_1",
sample = lambda wildcards: "03-stacks-analysis/de-novo/" wildcards.sample
output:
"03-stacks-analysis/de-novo/{sample}.matches.tsv.gz"
log: "03-stacks-analysis/de-novo/{sample}.sstacks.oe"
shell:"""
sstacks -c {params.catalog} -s {params.sample} -o 03-stacks-analysis/de-novo &> {log}
"""
# reference-based data analysis
## read alignment with bwa-mem
rule bwa_mem:
input: "01-clean-data/{sample}.fq.gz"
params:
index = INDEX,
mismatch = "3",
gap = "5,5"
threads: 8
output: "02-read-alignment/ref-based/{sample}.bam"
shell:"""
mkdir -p 02-read-alignment
bwa mem -t {threads} -M -B {params.mismatch} -O {params.gap} {params.index} {input} \
| samtools view -b > {output}
"""
## find variant loci
rule pstacks:
input: "02-read-alignment/ref-based/{sample}.bam"
params:
outdir = "03-stacks-analysis/ref-based/",
index = lambda wildcards: INDEX_DICT.get(wildcards.sample)
threads: 8
output:
"03-stacks-analysis/ref-based/{sample}.snps.tsv.gz",
"03-stacks-analysis/ref-based/{sample}.tags.tsv.gz",
"03-stacks-analysis/ref-based/{sample}.models.tsv.gz",
"03-stacks-analysis/ref-based/{sample}.alleles.tsv.gz"
log: "03-stacks-analysis/ref-based/{sample}.pstacks.oe"
shell:"""
mkdir -p 03-stacks-analysis/ref-based
pstacks -p {threads} -t bam -f {input} -i {params.index} -o {params.outdir} &> {log}
"""
## A catalog can be built from any set of samples processed by the ustacks or pstacks programs
rule ref_based_cstacks:
input: "info/popmap.tsv",expand("03-stacks-analysis/ref-based/{sample}.snps.tsv.gz", sample=SAMPLES)
threads: 10
output:
"03-stacks-analysis/ref-based/batch_1.catalog.alleles.tsv.gz",
"03-stacks-analysis/ref-based/batch_1.catalog.snps.tsv.gz",
"03-stacks-analysis/ref-based/batch_1.catalog.tags.tsv.gz"
shell:
"cstacks -p {threads} --aligned -P 03-stacks-analysis/ref-based/ -M {input[0]}"
## Sets of stacks, i.e. putative loci, constructed by the ustacks or pstacks programs
## can be searched against a catalog produced by cstacks.
rule ref_based_sstacks:
input:
"03-stacks-analysis/ref-based/{sample}.snps.tsv.gz",
"03-stacks-analysis/ref-based/{sample}.tags.tsv.gz",
"03-stacks-analysis/ref-based/{sample}.models.tsv.gz",
"03-stacks-analysis/ref-based/{sample}.alleles.tsv.gz"
params:
catalog = "03-stacks-analysis/ref-based/batch_1",
sample = lambda wildcards: "03-stacks-analysis/ref-based/" wildcards.sample
output:
"03-stacks-analysis/ref-based/{sample}.matches.tsv.gz"
log: "03-stacks-analysis/ref-based/{sample}.sstacks.oe"
shell:"""
sstacks --aligned -c {params.catalog} -s {params.sample} -o 03-stacks-analysis/ref-based &> {log}
"""将以上代码保存为Snakefile,没有集群服务器,就直接用 snakemake运行吧。因为我能在集群服务器上提交任务,所以用如下代码
snakemake --cluster "qsub -V -cwd" -j 20 --local-cores 10 &
plot_n_snps_per_locus.R的代码
#!/usr/bin/env Rscript
snps_per_loc = read.delim(\'./n_snps_per_locus.tsv\')
# Keep only M==n, m==3
snps_per_loc = subset(snps_per_loc, M==n & m==3)
# Rename column 1
colnames(snps_per_loc)[1] = \'par_set\'
# Create a new data frame to contain the number of loci and polymorphic loci
d = snps_per_loc[,c(\'par_set\', \'M\', \'n\', \'m\')]
d = d[!duplicated(d),]
# Compute these numbers for each parameter set, using the par_set column as an ID
rownames(d) = d$par_set
for(p in rownames(d)) {
s = subset(snps_per_loc, par_set == p)
d[p,\'n_loci\'] = sum(s$n_loci)
s2 = subset(s, n_snps > 0)
d[p,\'n_loci_poly\'] = sum(s2$n_loci)
}
# Make sure the table is ordered
d = d[order(d$M),]
pdf(\'./n_loci_Mn.pdf\')
# Number of loci
# ==========
plot(NULL,
xlim=range(d$M),
ylim=range(c(0, d$n_loci)),
xlab=\'M==n\',
ylab=\'Number of loci\',
main=\'Number of 80%-samples loci as M=n increases\',
xaxt=\'n\',
las=2
)
abline(h=0:20*5000, lty=\'dotted\', col=\'grey50\')
axis(1, at=c(1,3,5,7,9))
legend(\'bottomright\', c(\'All loci\', \'Polymorphic loci\'), lty=c(\'solid\', \'dashed\'))
lines(d$M, d$n_loci)
points(d$M, d$n_loci, cex=0.5)
lines(d$M, d$n_loci_poly, lty=\'dashed\')
points(d$M, d$n_loci_poly, cex=0.5)
# Number of new loci at each step (slope of the previous)
# ==========
# Record the number of new loci at each parameter step
d$new_loci = d$n_loci - c(NA, d$n_loci)[1:nrow(d)]
d$new_loci_poly = d$n_loci_poly - c(NA, d$n_loci_poly)[1:nrow(d)]
# Record the step size
d$step_size = d$M - c(NA, d$M)[1:(nrow(d))]
plot(NULL,
xlim=range(d$M),
ylim=range(c(0, d$new_loci, d$new_loci_poly), na.rm=T),
xlab=\'M==n\',
ylab=\'Number of new loci / step_size (slope)\',
main=\'Number of new 80%-samples loci as M=n increases\'
)
abline(h=0, lty=\'dotted\', col=\'grey50\')
legend(\'topright\', c(\'All loci\', \'Polymorphic loci\'), lty=c(\'solid\', \'dashed\'))
lines(d$M, d$new_loci / d$step_size)
points(d$M, d$new_loci / d$step_size, cex=0.5)
lines(d$M, d$new_loci_poly / d$step_size, lty=\'dashed\')
points(d$M, d$new_loci_poly / d$step_size, cex=0.5)
null=dev.off()
plot_n_loci.R的代码
#!/usr/bin/env Rscript
d = read.delim(\'./n_snps_per_locus.tsv\')
# Keep only M==n, m==3.
d = subset(d, M==n & m==3)
# Make sure the table is ordered by number of snps.
d = d[order(d$n_snps),]
Mn_values = sort(unique(d$M))
# Write the counts in a matrix.
m = matrix(NA, nrow=length(Mn_values), ncol=max(d$n_snps) 1)
for(i in 1:nrow(d)) {
m[d$M[i],d$n_snps[i] 1] = d$n_loci[i] # [n_snps 1] as column 1 is for loci with 0 SNPs
}
# Truncate the distributions.
max_n_snps = 10
m[,max_n_snps 2] = rowSums(m[,(max_n_snps 2):ncol(m)], na.rm=T)
m = m[,1:(max_n_snps 2)]
m = m / rowSums(m, na.rm=T)
# Draw the barplot.
pdf(\'n_snps_per_locus.pdf\')
clr = rev(heat.colors(length(Mn_values)))
barplot(m,
beside=T, col=clr, las=1,
names.arg=c(0:max_n_snps, paste(\'>\', max_n_snps, sep=\'\')),
xlab=\'Number of SNPs\',
ylab=\'Percentage of loci\',
main=\'Distributions of the number of SNPs per locus\nfor a range of M==n values\'
)
legend(\'topright\', legend=c(\'M==n\', Mn_values), fill=c(NA, clr))
null=dev.off()第三步:过滤并导出数据
这一步的过滤在stacks1.47是分为两个部分。第一部分是对于从头分析和基于参考基因组都使用rxstacks过滤掉低质量变异,然后重新用cstacks和sstacks处理。第二部分是使用population从群体角度进行过滤。 在stacks2.0时代,rxstacks功能不见了(我推测是作者认为作用不大),既然他们都不要了,那我也就只用population过滤和导出数据了。
这一步主要淘汰那些生物学上不太合理,统计学上不显著的位点,一共有四个主要参数负责控制,前两个是生物学控制,根据研究主题而定
- (-r):该位点在单个群体的所有个体中的最低比例
- (-p): 该位点至少需要在几个群体中存在
- (—min_maf): 过滤过低频率位点,推荐5~10%
- (—max_obs_het): 过滤过高杂合率位点, 推荐60~70%
# 项目根目录
min_samples=0.80min_maf=0.05max_obs_het=0.70populations -P 03-stacks-analysis/ref-based/ -r $min_samples --min_maf $min_maf \
--max_obs_het $max_obs_het --genepop &> populations.oe
# --genepop表示输出格式,可以选择--vcf等最后会在03-stacks-analysis/ref-based/生成至少如下几个文件:
batch_1.sumstats.tsv: 核酸水平上的描述性统计值,如观测值和期望的杂合度 π, and FISbatch_1.sumstats_summary.tsv: 每个群体的均值batch_1.hapstats.tsv:单倍型水平的统计值,如基因多样性和单倍型多样性batch_1.haplotypes.tsv: 单倍型batch_1.genepop:指定的输出格式batch_1.populations.log:输出日志
至此,上游分析结束,后续就是下游分析。后续更新计划:
- stacks总体流程鸟瞰
- Stacks核心参数深入了解
- RAD-seq和GBS技术比较
- 不同简化基因组protocol的比较
- 学习TASSEL-GBS数据分析流程
- 下游分析探索:这个得慢慢来