

看了illumina的测序仪市场份额的确很夸张,像我这样在生信数据分析领域身经百战的老鸟,都是直到今天才碰到color space的测序数据。测序平台是AB 5500xl Genetic Analyzer,就是传说中的solid格式。主要是我在学习一篇关于tp53转录因子结合能力的文章的时候碰到的 ,我查看了下载的数据虽然还是fastq格式,但很诡异,我完全不认识里面的序列。这里总结一下,下面是我的学习过程及思路,有点乱,大家随便看看!
首先:测序仪给的数据应该是 (.csfasta & .qual) 这两个后缀名的文件
然后,可以用脚本把数据转为csfastq格式, 与普通fastq数据格式是没有区别,但是里面包含的不是序列,是color的编码。
其次,color space不允许转为base space数据!!!
最后,之所以转为csfastq格式,是为了适应很多软件,fastqc,cutadap,SHRiMP,sequel和BFAST ,bowtie等等
csfastq数据如下,还是四行代表一条read:
@SRR2967009.1 100_1000_1168_F3
T10011023211201220121202030102221012302121010131001
2@@@@>@?@@@@<@@//;@@/@9?@8@=@@@6;6@66;<@6@67?2?;/@
@SRR2967009.2 100_1000_1211_F3
T20132312201120021312220200023110220113100012321011
@@@@@@@@@<@@@@@@@@@@@@@@@@@@@@@@?@@@@/?@@@@@@@@
比如对http://www.ncbi.nlm.nih.gov/sra?term=SRP066824 来说:
首先下载数据并且解压:
for ((i=7009;i<7014;i )) ;do wget ftp://ftp-trace.ncbi.nlm.nih.gov/sra/sra-instant/reads/ByStudy/sra/SRP/SRP066/SRP066824/SRR296$i/SRR296$i.sra;done
因为测序平台是AB 5500xl Genetic Analyzer,就是传说中的solid格式,所以不应该用fastq-dump啦,应该用abi-dump才对!
参考:http://davetang.org/muse/2012/07/04/from-sra-to-fastq-for-solid-data/
ls *sra |while read id; do ~/biosoft/sratoolkit/sratoolkit.2.6.3-centos_linux64/bin/abi-dump $id;done
解压之后是下面这样:
这样只能转为csfasta格式文件和qual文件,需要下载大名鼎鼎的lh写的一个脚本:wget http://www.bbmriwiki.nl/svn/bwa_45_patched/solid2fastq.pl 来转为fastq格式
程序非常好用:perl solid2fastq.pl SRR2967009_ SRR2967009 即可
也可以用Python程序来做这个转换,http://edison.cremag.org/resources/seq-analysis/tools/solid2fastq/
最后就是输出了fastq格式的 color space的数据 ,但是我测试了,直接用fastq-dump也可以把数据解压成fastq格式的color space的数据,并不需要那么麻烦的, 因为我们不是从测序仪拿数据,而是从SRA数据库里面直接下载。(补充一下,直接用fastq-dump也可以把数据解压成fastq格式跟用abi-dump解压后再转换成csfastq有区别,但是我现在说不清楚区别是什么,建议用abi-dump)
SOLiD native (CSFASTA/QUAL)
All SRA data can be output into color space data. The utility ‘abi-dump’ can be used to output CSFASTA and QUAL data files (with appropriate options, fastq-dump can be used to output “CSFASTQ” format).
SHRiMP,sequel和BFAST 都可以来比对fastq格式的color space的数据,或者直接从 (.csfasta & .qual) 这两个文件开始处理,其实bowtie也可以的。
https://wikis.utexas.edu/display/bioiteam/BFAST
比对后的bam文件,就可以走正常的illumina数据分析流程啦!
转为了fastq格式的color space的数据,就可以直接进行fastqc看看质量控制图片,如果质量很差,可以直接用处理cutadapt等各种软件进行处理,in a .csfasta and a .qual file (this is the native SOLiD format).
参考:http://cutadapt.readthedocs.io/en/stable/colorspace.html
fastqc软件直接处理csfastq格式数据结果如下:
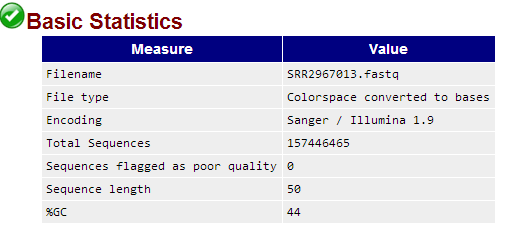
参考:http://seqanswers.com/forums/showpost.php?p=59156&postcount=4
Sequencer reads have a chance of read error (e.g. spot misidentification), combined with a chance of sequence error (e.g. polymerase misread in the PCR step).
For sequencers that output in base space, both these errors have a similar effect on the base-space mapping.
For sequencers that output in color-space, the read errors result in a somewhat unexpected base-space translation even if the underlying sequence has a perfect match to the reference.
The issues relating to color-space to base-space translation were discussed in the thread you linked to, but here’s my take on it (dumped from an email I recently sent to someone else):A color-space sequence is an encoding of adjacent dimers such that unchanging bases are encoded with ‘0’, complementary changes are encoded with ‘3’, the colour ‘1’ is used for a non-complementary base change on the same side of the alphabet (AC, CA, GT, or TG), and the colour ‘2’ is used for a non-complementary base change on a different side of the alphabet (AG, GA, CT, or TC). A table of these changes can be found here:
http://www.ploscompbiol.org/article/…i.1000386.g002
This has a few nice properties (e.g. the reverse-complement of a color-space sequence is the same as the reverse of the color-space sequence, a SNP will have two transitions), but many annoying and nasty properties.
The first is that a color-space sequence in itself is meaningless without a base reference (usually the starting base).
原文来自:http://www.bio-info-trainee.com/1850.html