

Harmony原理
Harmony需要输入低维空间的坐标值(embedding),一般使用PCA的降维结果。Harmony导入PCA的降维数据后,会采用soft k-means clustering算法将细胞聚类。常用的聚类算法仅考虑细胞在低维空间的距离,但是soft clustering算法会考虑我们提供的校正因素。这就好比我们的高考加分制度,小明高考成绩本来达不到A大学的录取分数线,但是他有一项省级竞赛一等奖加10分就够线了。同样的道理,细胞c2距离cluster1有点远,本来不能算作cluster1的一份子;但是c2和cluster1的细胞来自不同的数据集,因为我们期望不同的数据集融合,所以破例让它加入cluster1了。聚类之后先计算每个cluster内各个数据集的细胞的中心点,然后根据这些中心点计算各个cluster的中心点。最后通过算法让cluster内的细胞向中心聚集,实在收敛不了的离群细胞就过滤掉。调整之后的数据重复:聚类—计算cluster中心点—收敛细胞—聚类的过程,不断迭代直至聚类效果趋于稳定。
官方图解及正式说明
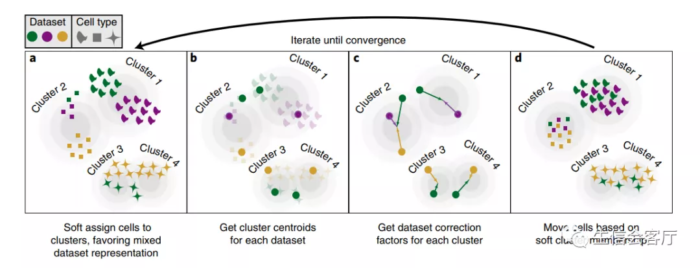
Overview of Harmony algorithm. PCA embeds cells into a space with reduced dimensionality. Harmony accepts the cell coordinates in this reduced space and runs an iterative algorithm to adjust for dataset specific effects.
a, Harmony uses fuzzy clustering to assign each cell to multiple clusters, while a penalty term ensures that the diversity of datasets within each cluster is maximized. b, Harmony calculates a global centroid for each cluster, as well as dataset-specific centroids for each cluster. c, Within each cluster, Harmony calculates a correction factor for each dataset based on the centroids. d, Finally, Harmony corrects each cell with a cell-specific factor: a linear combination of dataset correction factors weighted by the cell’s soft cluster assignments made in step a. Harmony repeats steps a to d until convergence. The dependence between cluster assignment and dataset diminishes with each round. Datasets are represented with colors, cell types with different shapes.
Harmony性能评估
作者使用harmony整合细胞系样本,HCA计划的免疫细胞大数据集,不同试剂的10X PBMC数据,不同技术平台的胰岛细胞数据,时间序列的小鼠发育样本数据,以及单细胞数据与空间转录组数据,效果都非常理想,不理想的数据也不会放出来。真正让我心动的是它极大的节省了计算资源,50万个细胞的整合只需要一个小时,内存占用也只有7.2G,如下图所示:
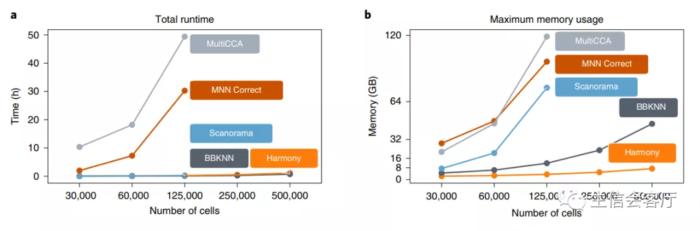
为了证实harmony的效果是否真的如作者说的那么好,我查到了一篇14种单细胞数据批次校正工具对比的文献,第三方横向对比测试也证实了harmony真的又快又好!有图为证:
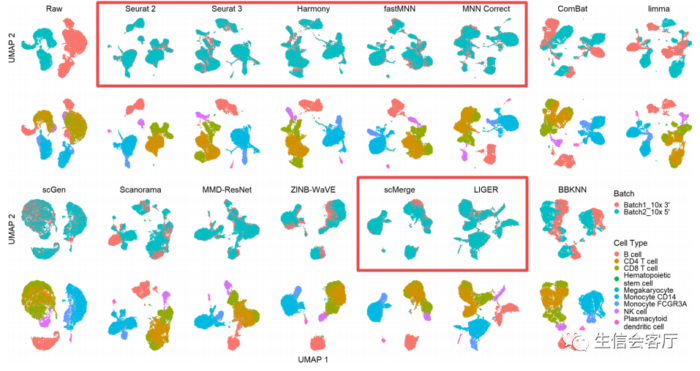
红色方框选择的是批次效应校正比较好的方法,作者比较了多个场景的数据整合效果,这里展示的只是其中一个场景。下面的A、B、C图展示的是14种工具最终的评比结果。图A是总体性能评分,底部是性能好的;图B是内存消耗对比,图C是运行时间对比。
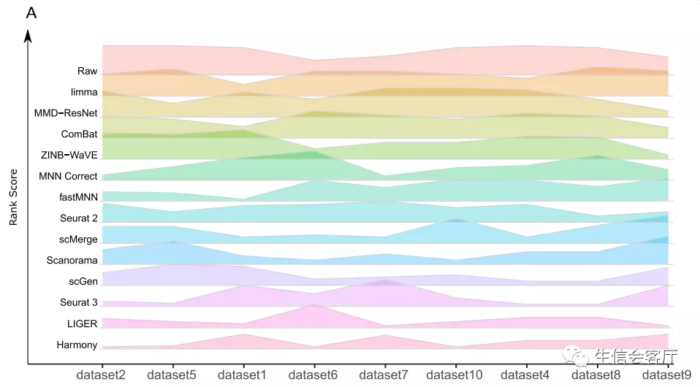
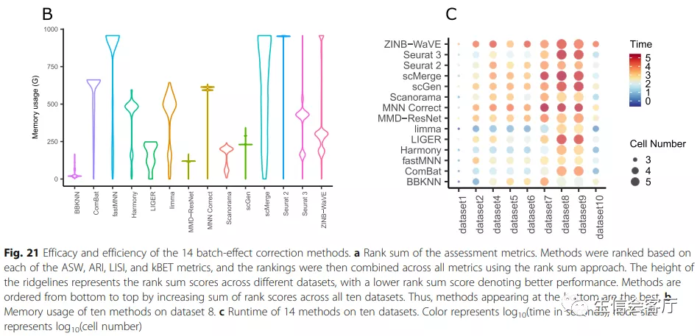
作者结论:Based on our results, we found LIGER, Harmony, and Seurat3 to be the top batch mixing methods. 从第三方的评测结果来看,harmony的整合效果可以与seurat的锚点整合方法媲美,并且运行时间方面具有明显优势,不过内存消耗好像没有那么夸张的优秀。
10X数据实测
安装harmony
#此包安装简单,没有特殊的依赖包
library(devtools)
install_github("immunogenomics/harmony")测试数据准备
测试数据采用《单细胞转录组分析:多样本合并与批次校正》一文中使用的那10个样本的数据,没有数据的朋友可以添加我的微信后索取,微信二维码在原文可以找到。
##==准备seurat对象列表==##
library(Seurat)
library(tidyverse)
library(patchwork)
rm(list=ls())
setwd("~/project/harmony")
dir <- dir("GSE139324/") #GSE139324是存放数据的目录
dir <- paste0("GSE139324/",dir)
sample_name <- c('HNC01PBMC', 'HNC01TIL', 'HNC10PBMC', 'HNC10TIL', 'HNC20PBMC',
'HNC20TIL', 'PBMC1', 'PBMC2', 'Tonsil1', 'Tonsil2')
scRNAlist <- list()
for(i in 1:length(dir)){
counts <- Read10X(data.dir = dir[i])
scRNAlist[[i]] <- CreateSeuratObject(counts, project=sample_name[i], min.cells=3, min.features = 200)
scRNAlist[[i]][["percent.mt"]] <- PercentageFeatureSet(scRNAlist[[i]], pattern = "^MT-")
scRNAlist[[i]] <- subset(scRNAlist[[i]], subset = percent.mt < 10)
}
saveRDS(scRNAlist, "scRNAlist.rds")下面我将分别使用Seurat和harmony整合数据,并统计时间和内存消耗。
Seurat整合样本
##==seurat整合多样本==##
scRNAlist <- readRDS("scRNAlist.rds")
for (i in 1:length(scRNAlist)) {
scRNAlist[[i]] <- NormalizeData(scRNAlist[[i]])
scRNAlist[[i]] <- FindVariableFeatures(scRNAlist[[i]]
}
##以VariableFeatures为基础寻找锚点
system.time({scRNA.anchors <- FindIntegrationAnchors(object.list = scRNAlist)})
# 用户 系统 流逝
# 605.661 4.949 610.328
##利用锚点整合数据,运行时间较长
system.time({scRNA_seurat <- IntegrateData(anchorset = scRNA.anchors)})
# 用户 系统 流逝
# 171.598 18.471 189.932
scRNA_seurat <- ScaleData(scRNA_seurat) %>% RunPCA(verbose=FALSE)
scRNA_seurat <- RunUMAP(scRNA_seurat, dims = 1:30)
scRNA_seurat <- FindNeighbors(scRNA_seurat, dims = 1:30) %>% FindClusters()
##作图
#group_by_cluster
plot1 = DimPlot(scRNA_seurat, reduction = "umap", label=T)
#group_by_sample
plot2 = DimPlot(scRNA_seurat, reduction = "umap", group.by='orig.ident')
#combinate
plotc <- plot1 plot2
ggsave("scRNA_seurat.png", plot = plotc, width = 10, height = 5)
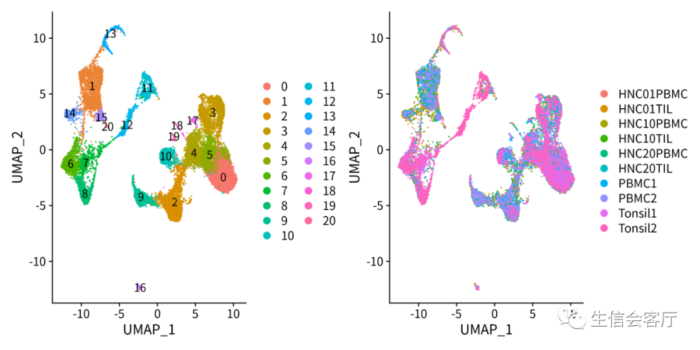
saveRDS(scRNA_seurat, 'scRNA_seurat.rds')整合效果见下图:
Harmony整合样本
##==harmony整合多样本==##
library(harmony)
scRNAlist <- readRDS("scRNAlist.rds")
##PCA降维
scRNA_harmony <- merge(scRNAlist[[1]], y=c(scRNAlist[[2]], scRNAlist[[3]], scRNAlist[[4]], scRNAlist[[5]],
scRNAlist[[6]], scRNAlist[[7]], scRNAlist[[8]], scRNAlist[[9]], scRNAlist[[10]]))
scRNA_harmony <- NormalizeData(scRNA_harmony) %>% FindVariableFeatures() %>% ScaleData() %>% RunPCA(verbose=FALSE)
##整合
system.time({scRNA_harmony <- RunHarmony(scRNA_harmony, group.by.vars = "orig.ident")})
# 用户 系统 流逝
# 34.308 0.024 34.324
#降维聚类
scRNA_harmony <- RunUMAP(scRNA_harmony, reduction = "harmony", dims = 1:30)
scRNA_harmony <- FindNeighbors(scRNA_harmony, reduction = "harmony", dims = 1:30) %>% FindClusters()
##作图
#group_by_cluster
plot1 = DimPlot(scRNA_harmony, reduction = "umap", label=T)
#group_by_sample
plot2 = DimPlot(scRNA_harmony, reduction = "umap", group.by='orig.ident')
#combinate
plotc <- plot1 plot2
ggsave("scRNA_harmony.png", plot = plotc, width = 10, height = 5)
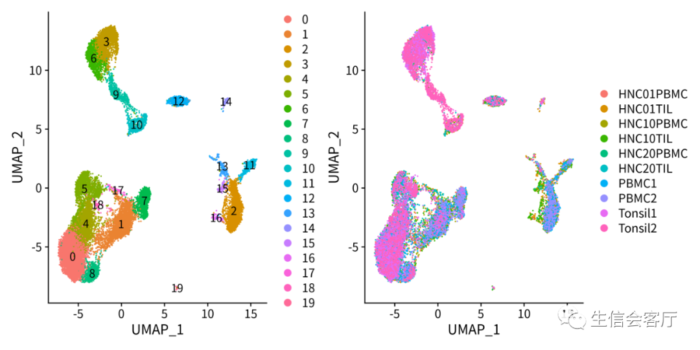
saveRDS(scRNA_harmony, 'scRNA_harmony.rds')整合效果见下图:
时间及内存消耗对比
本次整合的数据大约有2万个细胞,seurat整合步骤共用时800秒,harmony用时只有34秒。Seurat在整合的步骤可以支持并行计算,harmony没发现调整运行线程数的参数,应该不支持并行计算,不过seurat开启了并行计算模式速度也是不及harmony的。我记录了测试时间段的总内存(不包含buffers和cached的内存占用)使用情况:
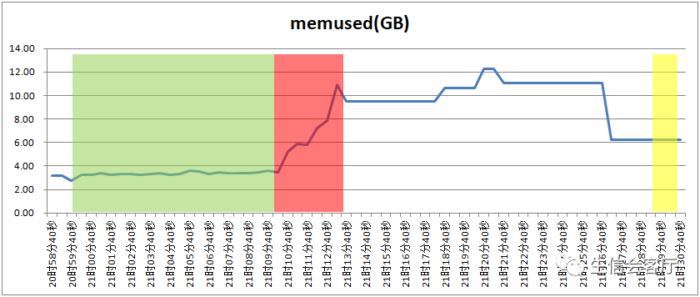
绿色部分是seurat运行FindIntegrationAnchors函数时间段,红色是seurat运行IntegrateData函数时间段,黄色是harmony运行RunHarmony函数的时间段。
分群效果评估
单纯从整合后降维图形看,很难判断哪个的效果更好。我准备等几个整合工具都介绍完了,统一用marker基因看看分群效果。