


【前言】其实这篇文章是为了简单介绍一下geneview的用法,它是一个Python高级库,建立在matplotlib的基础之上,专门用于基因组数据的可视化,目的是为了使创建高大上(精致)的基因组数据图表变得简单。目前该发布的Python包中已经内置多个优美的调色板和风格(默认情况下就能创建赏心悦目的图形),同时已经集成了曼哈顿图和Q-Q图的绘制函数。作为该Python包的主要开发者,只是如此是远远不够的,在未来的日子里,我希望它能在功能不断完善的同时也变得更加易用。
曼哈顿图和QQ图是两个在全基因组关联(GWAS)分析里面最常出现的图形,基本上已经是GWAS的标配,几乎在每篇GWAS的文章都会见到,它们的作用和所要传达出来的信息我也在[上一篇关于GWAS的博文]()中做了些说明,在这里我们就只集中在如何用Python和geneview将其有效地展现出来。
首先,准备一些数据来作为例子。
我这里用来展现的数据是2011年丹麦人所做过的一个关于年轻人过度肥胖的GWAS研究——GOYA,数据也是从他们所发表的结果中获得,总共有5,373个样本,其中超重的个体(case)有2,633个,正常的个体(control)是2,740个,从样本量上看还算可以。为了方便使用,我对其做了相关的处理,包括从PED和MAP文件到GEN文件的生成,并重复了一次case-control的关联性分析,计算出了芯片上所研究的各个SNP位点与肥胖相关的显著性程度(即p-value),最后又将结果数据抽取出来做成数据集——放在这里供下载(15.6Mb,csv格式)。
- 以上内容虽提及到了一些领域内术语和相关文件格式,但若不懂也请不必纠结,因为后续处理都是基于这个最终的数据集来完成的
接着,需要将geneview软件包加入到你的Python中,有多种不同的方式,但推荐直接使用pip,以下是安装比较稳定的发布版,直接在终端命令行下(Linux or Mac)输入:
pip install geneview
或者,也可以直接从github上安装正在开发的版本:
pip install git+git://github.com/ShujiaHuang/geneview.git#egg=geneview
第三种办法就是直接下载源码,然后自行编译,虽然不推荐这种做法(因为还有依赖包必须自行下载安装,过程会比较麻烦低效),但对于某些不能连接外网的集群也只能如此,这三种方式都是可行的。
曼哈顿图
将示例数据下载下来:
wget https://raw.githubusercontent.com/ShujiaHuang/geneview-data/master/GOYA.csv ./
先简单地查看一下数据的格式:
chrID,rsID,position,pvalue 1,rs3094315,742429,0.144586 1,rs3115860,743268,0.230022 1,rs12562034,758311,0.644366 1,rs12124819,766409,0.146269 1,rs4475691,836671,0.458197 1,rs28705211,890368,0.362731 1,rs13303118,908247,0.22912 1,rs9777703,918699,0.37948 1,rs3121567,933331,0.440824
一共是4列(逗号分隔),分别为:[1]染色体编号,[2]SNP rs 编号,[3] 位点在染色体上的位置,[4]显著性差异程度(pvalue)。在本例曼哈顿图中我们只需要使用第1,3和4列;而QQ图则只需要第4列——pvalue。
下面我们先从绘制曼哈顿图开始。我们先将需要的数据读取到一个列表中,可以这样做:
import csv data = [] with open(“GOYA.csv”) as f: f_csv = csv.reader(f) headers = next(f_csv) data = [[row[0], int(row[2]), float(row[3])] for row in f_csv]
现在GOYA.csv中的数据就都存放在data列表中了,由于Python在读取文件中数据时,都是以string类型存放,因此对于第3和第4列的数据有必要事先把做点类型转换。
接下来,调用geneview中的曼哈顿图函数。
import matplotlib.pyplot as plt from geneview.gwas import manhattanplot ax = manhattanplot(data, xlabel=”Chromosome”, ylabel=”-Log10(P-value)”) # 这就是Manhattan plot的函数 plt.show()
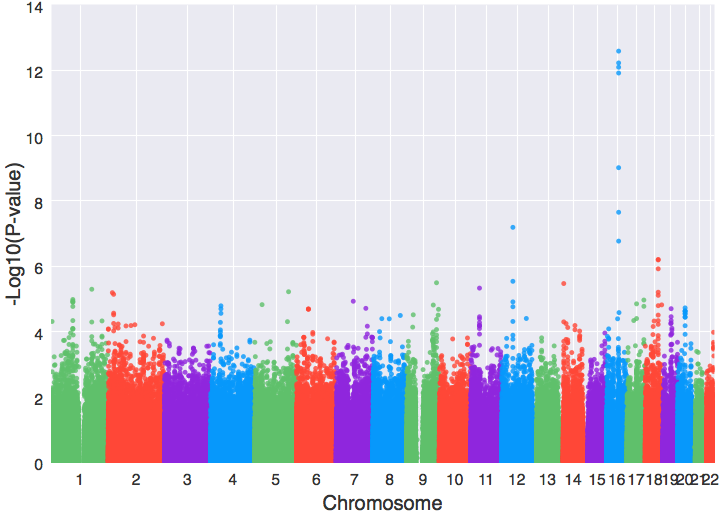
只需这样的一句代码就能创建一个漂亮的曼哈顿图,有必要再次指出的是,geneview是以matplotlib为基础开发出来的,所创建的图形对象实际上仍属于matplotlib,geneview内部自定义了很多图形风格,同时封装了大量只属于基因组数据的图表类型,但图形的输出格式以及界面显示都仍和matplotlib一样,因此在这里我们使用matplotlib.pyplot的show()函数(上例中:plt.show())将所绘制出来的曼哈顿图显示出来。如果要将图形保存下来,则只需执行`plt.savefig(“man.png”)`,这样就会在该目录下生成一个名为『man.png』png格式的曼哈顿图,若是要存为pdf格式,则只需将所要保存的文件名后缀改成『.pdf』(plt.savefig(“man.pdf”))就可以了。下面这些格式:emf, eps, pdf, png, jpg, ps, raw, rgba, svg, svgz等都是支持的,至于最新的还有多少种,还请参照matplotlib文档中说明。
此外,geneview中的每个画图函数都有着足够的灵活性,我们也可以根据自己的需要做一些调整,比如:
xtick = [‘1’, ‘2’,’3′,’4′,’5′,’6′,’7′,’8′,’9′,’10’,’11’,’12’,’13’,’14’,’16’,’18’, ’20’,’22’] manhattanplot(data, xlabel=”Chromosome”, # 设置x轴名字 ylabel=”-Log10(P-value)”, # 设置y轴名字 xtick_label_set = set(xtick), # 限定横坐标轴上的刻度显示 s=40, # 设置图中散点的大小 alpha=0.5, # 调整散点透明度 color=”#f28b1e,#9a0dea,#ea0dcc,#63b8ff”, # 设置新的颜色组合 )
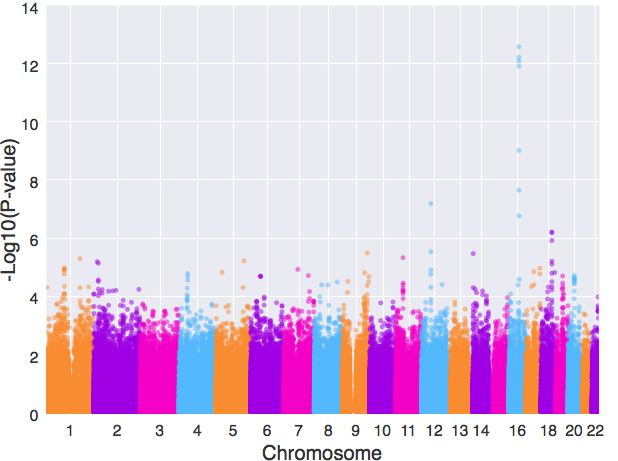
实现新的颜色组合、限定x轴上的刻度显示和散点大小的调节。甚至还可以将散点改为线:
manhattanplot(data, xlabel=”Chromosome”, # 设置x轴名字 ylabel=”-Log10(P-value)”, # 设置y轴名字 xtick_label_set = set(xtick), # 限定横坐标轴上的刻度显示 alpha=0.5, # 调整散点透明度 color=”#f28b1e,#9a0dea,#ea0dcc,#63b8ff”, # 设置新的颜色组合 kind=”line” )
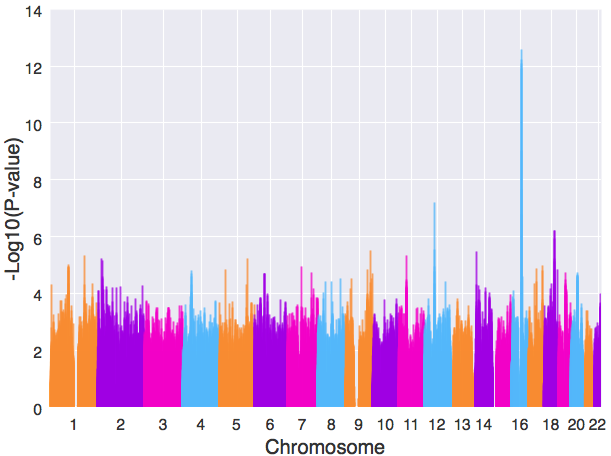
其它方面的调整请查看geneview文档中的相关说明。
Q-Q图
qq图只需用到上例中的pvalue那一列:
import csv import matplotlib.pyplot as plt from geneview.gwas import qqplot pvalue=[] with open(“GOYA.csv”) as f: f_csv = csv.reader(f) headers = next(f_csv) pvalue = [float(row[3]) for row in f_csv] ax = qqplot(pvalue, color=”#00bb33″, xlabel=”Expected p-value(-log10)”, ylabel=”Observed p-value(-log10)”) # Q-Q 图 plt.show()
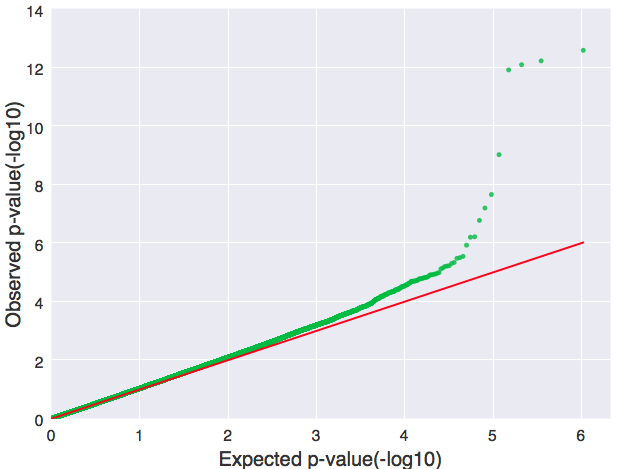
同样,也可以根据自己的需要对改图进行相关的调整。
以上,便是如何使用Python来制作Manhattan图和QQ图的方法,geneview的集成函数简化了这样的一个过程。
另外,如果你也看过丹麦人的这个GOYA研究,就会发现实际以上的两个图和其文章中的基本是一致的,当然我自己做了
些数据清洗的操作,结果上仍然会有些许的不同。虽然此刻下结论还有点为时尚早,但总的来讲,我应该也可以通过这个数据集比较顺利的将其结果重复出来了。
最后,附上利用geneview画曼哈顿图和QQ图的代码:
(1)曼哈顿图:
import sys
import csv
import matplotlib.pyplot as plt
sys.path.append('..')
import geneview as gv
xtick = ['chr'+c for c in map(str, range(1, 15) + ['16', '18', '20', '22'])]
df = gv.util.load_dataset('GOYA_preview')
gv.gwas.manhattanplot(df[['chrID','position','pvalue']],
hline_kws={'y': 3, 'color': 'b', 'lw': 1},
xlabel="Chromosome",
ylabel="-Log10(P-value)",
xticklabel_kws={'rotation': 'vertical'},
xtick_label_set = set(xtick))
plt.savefig('manhattan.png')
plt.show()(2)QQ图:
import sys
import matplotlib.pyplot as plt
sys.path.append('..')
import geneview as gv
df = gv.util.load_dataset('GOYA_preview')
gv.gwas.qqplot(df['pvalue'])
"""
fig, ax = plt.subplots()
gv.gwas.qqplot(df['pvalue'], ax=ax,
xlabel="Expected p-value(-log10)",
ylabel="Observed p-value(-log10)")
"""
plt.savefig('qq.png')
plt.show()
1F
您好, 谢谢你的分享。
我刚开始学python,对这篇文章很感兴趣,所以按照你写的代码亲自尝试了下。 但是在运行中出现了一点问题, 希望你能帮助我,非常感谢。
import matplotlib.pyplot as plt
from geneview.gwas import manhattanplot
ax = manhattanplot(data, xlabel=’Chromosome’, ylabel=’-Log10(P-value)’) # 这就是Manhattan plot的函数
plt.show()
我在运行以上代码时, 得到了下面的错误信息。不知道您有什么方法可以调试下。
—————————————————————————
AttributeError Traceback (most recent call last)
in ()
3 from geneview.gwas import manhattanplot
4
—-> 5 ax = manhattanplot(data, xlabel=’Chromosome’, ylabel=’-Log10(P-value)’) # 这就是Manhattan plot的函数
6 plt.show()
/anaconda3/lib/python3.7/site-packages/geneview/gwas/_manhattan.py in manhattanplot(data, ax, xlabel, ylabel, color, kind, xtick_label_set, CHR, alpha, mlog10, **kwargs)
141 if CHR is not None and seqid != CHR: continue
142
–> 143 color = colors.next()
144 region_xs = [last_x + r for r in rlist[‘pos’]]
145 x.extend(region_xs)
AttributeError: ‘itertools.cycle’ object has no attribute ‘next’
B1
@ tuibian 一样遇到一楼的问题,求解答!
源码:
import matplotlib.pyplot as plt
import geneview as gv
import csv
data=[]
with open(‘information.csv’) as f:
f_csv=csv.reader(f)
headers=next(f_csv)
data=[[row[0],int(row[2]),float(row[3])]for row in f_csv]
xtick=[‘1′,’2′,’3’]
ax=gv.gwas.manhattanplot(data,xlabel=”Chromosome”,ylabel=”-Log10(P-value)”,s=10,alpha=0.5,xtick_label_set = set(xtick))
plt.show()
报错:
AttributeError: ‘itertools.cycle’ object has no attribute ‘next’
2F
有可以用于Windows的安装包吗?指点一下
3F
我用您在GitHub里的方法,不用csv而用DataFrame,但是仍然遇到相同的问题,热切期待您的解答!
源码:
import matplotlib.pyplot as plt
import geneview as gv
import pandas as pd
PandaRequest={‘chrID’:pd.Series([1,1,1,1,2,2,2,2,3]),
‘rsID’:pd.Series([‘rs3094315′,’rs3115860′,’rs12562034′,’rs12124819′,’rs4475691′,’rs28705211′,’rs13303118′,’rs9777703′,’rs3121567’]),
‘position’:pd.Series([742429,743268,758311,766409,836671,890368,908247,933331]),
‘pvalue’:pd.Series([0.144586,0.230022,0.644366,0.146269,0.458197,0.362731,0.22912,0.37948,0.440824])}
data=pd.DataFrame(PandaRequest)
xtick=[‘1′,’2′,’3’]
gv.gwas.manhattanplot(data[[‘chrID’,’position’,’pvalue’]],
xlabel=”Chromosome”,
ylabel=”-Log10(P-value)”,
s=10,
alpha=0.5,
xtick_label_set = set(xtick))
plt.show()
报错:
AttributeError: ‘itertools.cycle’ object has no attribute ‘next’