

# 用awk进行简单的编程
# 我们之前已经做过这个了,但是为了让大家我们了解我们正在做什么,我们来再做一遍。
# 提取埃博拉基因组的编码特征、基因以及编码序列。
efetch -db nucleotide -id NC_002549.1 -format gb > NC.gb
readseq -format=GFF -o NC.gff NC.gb
#*可以跳过上边两步,使用拟南芥gff文件。
#*curl -O ftp://ftp.arabidopsis.org/home/tair/Genes/TAIR10_genome_release/TAIR10_gff3/TAIR10_GFF3_genes.gff
#*我们可以先来查看一下gff格式是什么样子的
less -S TAIR10_GFF3_genes.gff |head
#*gff文件是tab分隔的文件。第1列是染色体信息;第2列是gff注释数据来源;第3列为特征(feature)即属于gene还是mRNA还是CDS等等;第4和5列分别是这个特征序列的起始和终止位置,第6列是得分,可以是序列相似性比对时的E-values值或者基因预测是的P-values值。”.”表示为空(来自参考资料1);第7列是表示序列的方向:正义链为+,反义链为-;第8列仅为对CDS的注释,表示起始编码的位置,有效值为0、1、2(来自参考资料1);第9列为注释信息。
# 找到每个特征的长度,注意“魔法”。
#*如果使用的是拟南芥的gff文件为操作对象,请自行将之后所有命令的NC.gff改为TAIR10_GFF3_genes.gff
#*$1, $2, $3指的就是第1-3列的数据
cat NC.gff | awk ' { print $1, $2, $3 } ' | head -5
# 这(基本上)等同于截取列。
cat NC.gff | cut -f 1,2,3 | head -5
# 我们可以进行计算。每个特征序列长度是多少?
#*第5列的数值减去第4列的数值后+1,即得到特征序列的长度
cat NC.gff | awk ' { print $3, $5-$4 + 1 } ' | head -5
# 我们可以利用模式匹配来提取CDS特征。
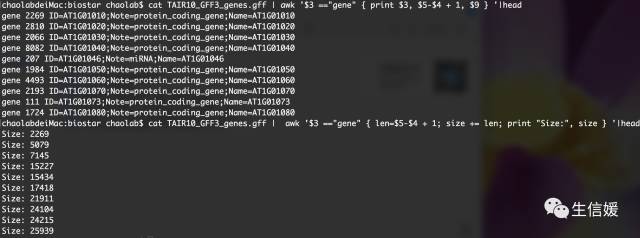
cat NC.gff | awk '$3 =="gene" { print $3, $5-$4 + 1, $9 } '
# 计算所有基因的累积长度。
cat NC.gff | awk '$3 =="gene" { len=$5-$4 + 1; size += len; print "Size:", size } '
# 计算所有CDS的累积长度。
cat NC.gff | awk '$3 =="CDS" { len=$5-$4 + 1; size += len; print "Size:", size } '
# 基因组有多大?有很多方法可以知道,比如从SAM文件的头文件(header)。
samtools view -h ../lec16/results.bam | head -2
# 基因组上有多少是基因。
#*如果是拟南芥,把perc=size/18959对应改成perc=size/119667750,119667750是拟南芥(Col-0)基因组的大小。
#*可以用下边的代码自行计算。
#*cat TAIR10_GFF3_genes.gff |awk '$3 == "chromosome"{len=$5-$4 + 1; size += len; print "Size:", size } '
cat NC.gff | awk '$3 =="CDS" { len=$5-$4 + 1; size += len; perc=size/18959; print "Size:", size, perc } '
# 我们需要使awk只用tab分隔符进行分隔。
# 用空格符分隔虽然有用,但是常常导致烦人的bugs
# 告诉awk,以tab作为F(字段分隔符)和OFS(输出字段分隔符)
# 把下边这行命令放到 .profile 或 .bashrc 文件中
alias awk="awk -F '\t' -v OFS='\t'"
# 使设置生效。
source ~/.profile
# 生成新的含有基因和CDS的gff文件。
# The first line specifies what kind of file this is.
head -1 NC.gff > NC-genes.gff
head -1 NC.gff > NC-cds.gff
# 根据特征(features)把文件分开。
cat NC.gff | awk ' $3=="gene" { print $0 }' >> NC-genes.gff
cat NC.gff | awk ' $3=="CDS" { print $0 }' >> NC-cds.gff
# Sam文件是tab分隔符的,可以用awk来处理。
# 上周的数据中,有多少碱基被覆盖了超过50次?
samtools depth ../lec16/results.bam | awk '$3 > 50 { print $0 } ' | wc -l
# 有多少的模板长度超过50 bp。
samtools view ../lec16/results.bam | awk ' $9 > 50 { print $0 } ' | wc -l
从GFF文件中分离提取基因名字。
$3 == "gene" {
# 通过 ; 分离提取第9列
split($9, x, ";")
# 基因名字是第一个元素。
# 通过空格符切分。基因名字是第二个元素
split(x[1], y, " ")
# 去除基因名字旁边的双引号
name = y[2]
#*对于拟南芥的gff,通过“=”切分。基因名字是第二个元素:
#*split(x[1], y, "=")
#*name = y[2]# 用空格全局替换 " 符号
# 由于 " 是一个特殊字符,我们必须写成 \"
#*反斜杠\表示转义符。
gsub("\"", "", name)
# 打印特征类型、基因名字以及大小。
print $3, name, $5 - $4 + 1}
#*最后,我们可以写成下边这条命令
#*cat TAIR10_GFF3_genes.gff |awk '$3 == "gene"{split($9,x,";");split(x[1], y, " ");name = y[2];gsub("\"", "", name); print $3, name, $5 - $4 + 1 } '