

DNA功能域(motif)简单地讲就是一段特定模式的DNA序列,它之所以可以具有生物学功能是因为它的特殊序列可以和调控蛋白结合,比如转录因子,从而可以在短暂时间内锚定功能蛋白。通常,DNA功能域的长度为5〜20bp,它可能出现在多个不同的基因附近,也可能在同一基因附近多次出现。它可以在双链中的任何一条上出现,因为转录因子是直接结合在DNA双链上的。DNA功能域被分成了两大类,一类是回文结构功能域(palindromic motifs),一类是二联体结构功能域(spaced dyad (gapped) motifs)。回文结构就是说无论正义链还是反义链都是一样的,比如CACGTG。而二联体结构是指在一小段序列的两边出现两个小的高度保守的序列,这两个高度保守的序列就是二联体,两中间的小段序列就称为空隔。空隔的出现,为二聚体(dimer)这样的转录因子提供了结合空间。通常,这个二联体的单体序列都很短,只有3~5bp。中间的空隔的长度基本固定,但是也可以较小的变化。
现在已经有大把的搜索算法来搜索DNA功能域。它们都有共同的前提假设,那就是人们所提供的输入序列是一些被相同转录因子调控的序列(coregulated genes)。因为只有这样,DNA功能域才会被富集起来,从而有可能从一大堆序列中发掘出来。然而我们知道,因为真核生物的表达调控的复杂性,所以这些算法大多都在原核生物中有较好的表现,甚至于酵母中中都有较好的表现,但到了其它真核生物中时,较很难有所作为。为了克服这一问题,人们使用了比较基因组学以及进化足迹(phylogenetic footprinting)等手段来进行调整,因为人们认为,在进化压力下,DNA功能域较其它非功能DNA序列相对保守许多。
功能域搜索算法
一般搜索对象被分为三大类,
- 同一基因组内的共调控基因(coregulated genes)的启动子区域
- 不同基因组间的同源基因的启动子区域
- 同源的共调控基因的启动子区域
而搜索算法主要被分成了两类,
- 字符搜索算法(word-based, string-based methods)
- 概率序列模型搜索算法(probabilistic sequence models)
字符搜索算法基于枚举序列,适合较短的功能域搜索,因为真核生物的DNA motif较原核的要短小,表面上看来它似乎较为有优势。但是因为大多数功能域其实还有空间上的选择,所以不得不加入后期的处理。而且,字符搜索会带来过多的侯选功能域。
概率序列模型搜索算法最终给出的结果通常都是一串位置权重矩阵(position weight matrix)。位置权重矩阵可以使用sequence logo来进行可视化显示,也就是由ATGC在每个位置所占比例而决定其高度堆叠显示其序列的图型。概率算法的优势在于所需要的参数少,但是因为它是基于模型的,所以不同的模型就可能带来分析结果的巨大差异。概率算法倾向于搜索更长,更为常见的功能域,所以它似乎更适合搜索原核生物的DNA功能域。由于抽样的问题,它们可能找到的并不是全局最佳解,而只是算法当中当前步骤的最佳解。
下面我们就只针对同一基因组内的共调控基因的启动子区域的DNA功能域搜索介绍。
字符搜索算法
van Helden是第一个把字符搜索算法应用到功能域搜索领域上来的人。尽管算法很简单,但是在搜索大多数酵母DNA功能域都非常有效。这一算法不但映证当时已经发现的很多DNA功能域,还准确地预测出了上游的一些功能域。相对于启发式的概率序列模型搜索算法,它是比较严密和全面的。但是它的搜索模式也相较的简单,只能针对包含高度保守的短序。后来van Helden又发展了可以搜索二联体结构功能域的算法。因为二联体结构功能域中间的空格长度可以会变化,所以在这一算法中,也设定空格的长度可以在0〜16之间摆动。而van Helden算法中最大的不足就是不提供碱基内的摆动。
Tompa发展了van Helden的算法。他的算法似乎有些更具有概率搜索的味道。Tompa使用z-score来对随机的指定长度的DNA序列进行排序从而得到DNA功能域。我们假设一段DNA序列在一条长度为L的片段里出现一次的概念为p,这个被称为背景。那么它在N条长度为L的片段里出现一次的概念就是Np。它的标准差为√Np(1-p)。那么z-score就是M=(N-Np)/√Np(1-p)。Tompa利用这一算法发展了YMF(Yeast Motif Finder)。它在研究中比较了YMF和MEME, AlignACE,认为YMF在酵母的DNA功能域的搜索中较另两者要准确。但是他也建议同时使用多种算法来分析同一数据,因为对于不同的数据,可能效果会不一样。
Brazma使用了正则匹配的办法来发展字符搜索算法。他搜索了两组酵母数据,一组是6000个推测基因的启动子区域,另一组是共调控基因的启动子区域。结果表明大部分搜索到的正测模板都得到了已知数据的支持。
还利用后缀树算法来进行字符搜索的。但是这些都是非主流。
概率算法
可能现在使用最多的就是概率算法了。其中使用最多的算法分为两类,一类是贪婪算法(greedy algorithm),包括其分支期望值最大化算法(expectation maximization, EM),另一类就是吉布斯取样(Gibbs sampling)。
EM算法
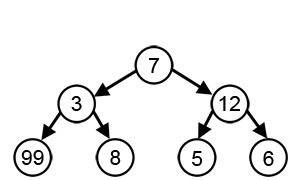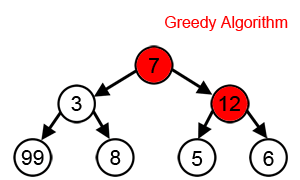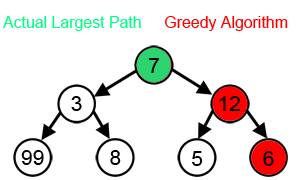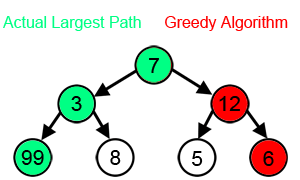
首先讲一下什么是贪婪算法,简单地讲,就是将一个大问题分成若干小的步骤,这些步骤是首尾相接的,每一步都要求最佳解,从而进入之后的步骤,最终将问题解决。如果一个问题可以使用贪婪算法取得答案,通常它都是最佳算法。但是使用贪婪算法解决的问题并不一定都严格适用贪婪算法,所以前一句等于白说。而DNA功能域搜索就是不严格适用的情况。如下图所示,每一步都希望找到最大值,从而得到总体相加的最大值,按照贪婪算法,那就会走7-12-6的路径,但实际上,7-3-99才是总体相加最大值。所以说,贪婪算法其实是一种马尔可夫链,前一步要决定下一步,而不是简单地下一步只是前一步的后续。而DNA功能域搜索就正好不严格符合贪婪算法的这个前提,但它毕竟大多数情况下可以拿到答案。
EM算法,多被用于聚类分析。比如下图。我们有一些观察数据,它是符合混合高斯分布的。我们现在的问题就是把这些点聚类分离开。假如我们用z来表示每一个高斯分布,那么这些数据就不仅仅是{x1,x2,x3…}了,它还有一个隐藏变量{(xi,zj),….}。因为我们不知道x属于哪个分布,所以z是我们观察不到的,z就被称为隐藏变量(latent variable)。虽然不知道z,但是我们可以用它的期望E去估计z。假设E值和三个变量π,μ,Σ有关,而如果π一但确定了,μ,Σ就可以通过观察到的x以及π来确定,从而得到E值。于是我们不断地的增长π值去更新E值,当E值达到最大时,我们从而得到估计的z值。而从一堆共调控序列中寻找功能域也可以类比这种聚类问题。只是分布函数不同罢了。
Hertz是第一个把贪婪算法引入的人,他期待找出在一堆共调控序列中都出现过的保守区域。后期这一算法得到了不断的发展。它的一个分枝EM算法是使用较多的。EM算法是被Lawrence和Reilly引入的。它一开始是做为蛋白功能域搜索而引入的,后来发现它在DNA功能域搜索方面也可以工作。后来由Bailey和Elkan发展出了大名鼎鼎的MEME算法。MEME算法的目标是搜索未知的功能域。它综合了之前很多的观点,首先,它使用实际存在于提供数据的短序做为EM算法的起点。其次,它摈弃了以前认为的每条序列中一个功能域只出现一次的假设。第三,认为同一条序列中可以含有多个功能域。而且,它的说明文档与维护工作做得非常不错,而且还开发出了大量相关的应用,所以它受到了许多生物学家的喜爱。
吉布斯取样算法
所谓吉布斯取样,简单地来讲,就是我们有几组变量,比如X,Y,Z。每一组当中的所有的枚举类型我们都知道,X={x1,x2,…,xm}, Y={y1,y2,…,yn}, Z={z1,z2,…,zw}。假设X,Y,Z有一定的关系,比如说选了x1就只能选Y的一部分子集,而选择某一个y,就只能选Z的一部分子集。于是随机选取一个出发点,xyz,而后不断地变更x,从x选取y,不断地变更y,从y选取z,从而得到一个xyz的集合。我们抛弃前面的一部分数据,而后固定间距取样,得到一个较小的子集,这就是吉布斯取样了。
而吉布斯取样如何应用到DNA功能域搜索中来呢?简单地,首先我们把所有的序列做个比对,得到一堆小的聚集的小片段。比如说这些类似的小片段有N个。我们就把包含这N个小片段的序列捆在一组。分组之后呢,再对组内使用吉布斯取样计算概率。它把组内每一条序列都当成一个变量,然后从中选取一个,把它切分成许多可能的功能域小片段(固定长度)做为它的变量集合,而后通过计算聚集的那个小片段的序列概率比较背景(整条序列的其它小片段概率)得出一个得分。之后是不停地抽取别的序列来变更这个小片段的概率得分。然后再把这件事情换个分组再做一遍,直到所有的聚集的小片段都搞定。
吉布斯取样和贪婪算法最大的不同就是它的取样看上去是随机的,并且符合指定的分布比如高斯分布。而不象贪婪算法,由前一步决定下一步的搜索目标,从而有可能避免贪婪算法中局部最优,但总体未必最优的缺点。但是明显,样品量的多少也会很大的影响概率分布与真值的接近程度。
Lawrence是使用吉布斯取样算法搜索功能域的第一人,他介绍了如果在功能域搜索上使用吉布斯取样,但是他只是使用了蛋白质的例子。基于这一结果,Roth发展出了AlignACE (Aligns Nucleic Acid Conserved Elements)算法。这一算法是针对DNA的。它与Lawrence他们的算法最大的不同就是,
- 因为对象是DNA,所以背景就不再是序列中的小片段了,而是固定为整个基因组的ATGC概率。
- 双链均会列入计算,但不允许overlapping。
- 如果某个功能域已经被确定,那么它会在之后的取样中剪除。
- 它使用了优化的取样算法。
它使用MAP(maximum a priori log-likelihood)来评价功能域的得分,但是MAP有个缺点,那就是如果这个片段在整个基因中出现的次数高的话,它的得分就非常高。但是这种片段往往并非功能域。后来Hughes等人在使用AlignACE时按组修正了MAP评分,使得它的表现更好。
Thijs发展出了MotifSampler算法。它对吉布斯取样进行了改进,两点,1是使用概率来估计功能域在同一条序列上出现的次数,2是使用了高阶马尔可夫链背景模型。他利用这一算法对拟兰介进行了DNA功能域的推测。
Liu分展出了BioProspector算法。它与原始的吉布斯取样不同的是:1,它使用了0〜3阶的马尔可夫链背景模型,参数可以手设,也可以自动获取。2,每个功能域的评分由monte Carlo方法估算。3,它重视回文结构和二联体结构。作者主要在原核生物中验证这一算法。
其它的算法还有诸如遗传算法,自组织神经网络,整数线性模型,以及基于结构的算法等。但大多非主流,就不介绍了,看得累。
Hu整了个大杂烩,叫EMD,包括了AlignACE, Bioprospector, MDScan, MEME以及MotifSampler。作者称大杂烩总比单个的好。但是,只有对每个算法软件都了解显然会在参数设置上才能上手。
算法综合评价
各个算法更有优势,但总体表现而言,都是假阳性过高。造成这一结果的原因很多,
- 调控机制知之甚少。
- 输入信噪比低。因为输入的都是一条条的序列,而功能域只是很短的一小节,其信噪比可想而知。更何况许多我们认为的共调控基因根本就不受相同的调控因子调控,这也增加了噪音。
- 其它
英文原文:
A survey of DNA motif finding algorithms, Modan K Das et. al., BMC Bioinformatics 2007, 8(suppl 7):S21 dio:10.1186/1471-2105-8-s7-s21 (http://www.biomedcentral.com/1471-2105/8/S7/S21/)
本文来自:http://pgfe.umassmed.edu/ou/archives/2355