

肿瘤是高度异质性的,其中致病突变可能以极低比例存在,比如allel frequency< 0.1%~0.01%,甚至更低。而目前最准的Hiseq测序本身的单个碱基的错误率就在0.2%左右,再加上众所周知的DNA聚合酶的固有错误率(10-7~10-5),那如何能测出0.1%的超低比例的突变?
MichaelW. Schmitta等人提出了极富创造力的方法,该方法的检测灵敏度达到0.01%,甚至更好。具体原理如下图:

(A) Adapter synthesis. A double-stranded, randomized Duplex Tag sequence is appended to a sequencing adapter by copying a degenerate sequence in one strand of the adapter with DNA polymerase. Complete adapter A-tailing is ensured by extended incubation with polymerase and dATP.
在Illumina的标准建库接头中的一条上加上12个简并碱基和4个固定序列碱基作为分子标签。
并通过复制把互补链也补上16个简并碱基。
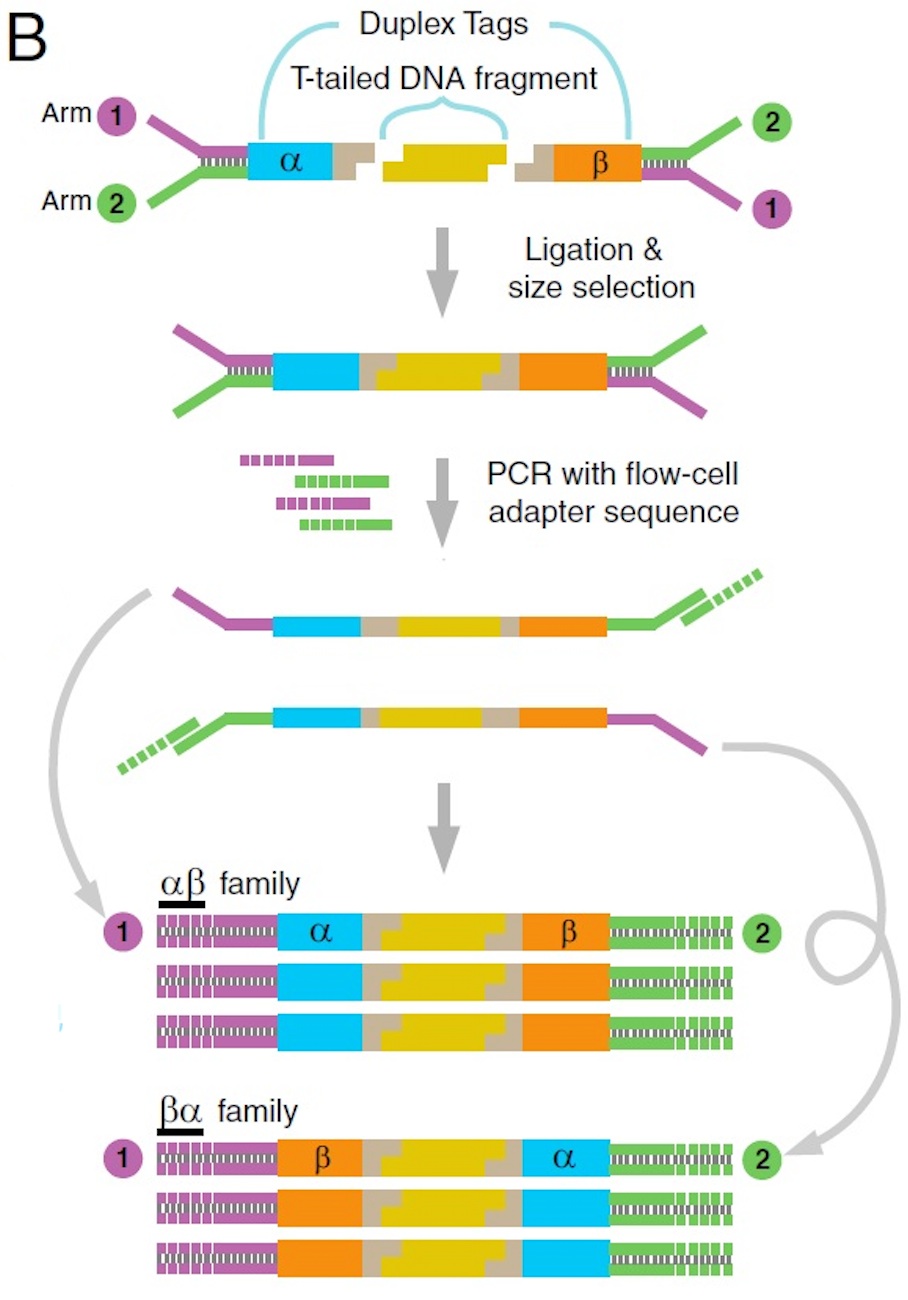
(B) Duplex Sequencing workflow. Sheared, T-tailed double-stranded DNA is ligated to A-tailed adapters. Because every adapter contains a Duplex Tag on each end, every DNA fragment becomes labeled with two distinct tag sequences (arbitrarily designated α and β in the single fragment shown). PCR amplification with primers containing Illumina flow-cell–compatible tails is carried out to generate families of PCR duplicates. Two types of PCR products are produced from each DNA fragment. Those derived from one strand will have the α tag sequence adjacent to flow cell sequence 1 and the β tag sequence adjacent to flow cell sequence 2. PCR products originating from the complementary strand are labeled reciprocally.
用改造好的接头与样本DNA进行连接,虽然接头总体上是简并的,但是具体到每个分子,还是有其特定的序列。
样本DNA加好接头后,得到的原始测序模板,而每个模板的每一头都被加上了12个碱基的分子标签,那每个模板的左、右两头加起来就有24个碱基的分子标签。
每个碱基是4种选择,24个碱基就是4的24次方,等于281T种可能性。这保证了每个原始模板在原始文库里都是独一无二的。
PCR扩增原始文库,每个模板会形成基于原始模板的2个中间序列互补的分子家族:正向和反向
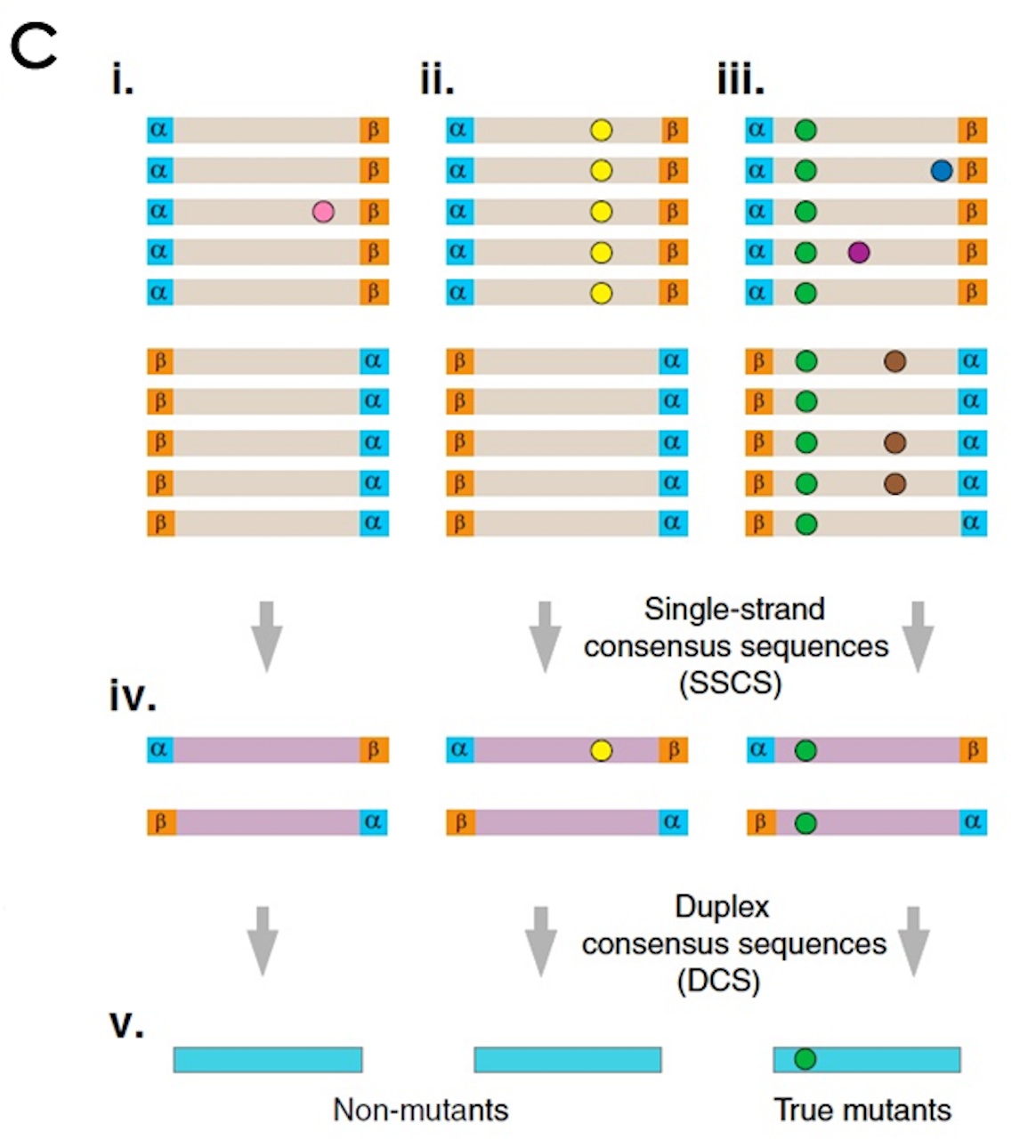
(C) Error correction. (i–iii) Sequence reads sharing a unique set of tags are grouped into paired families with members having strand identifiers in either the αβ or βα orientation. Each family pair reflects the amplification of one double-stranded DNA fragment. (i) Mutations (colored spots) present in only one or a few family members represent sequencing mistakes or PCR-introduced errors occurring late in amplification. (ii) Mutations occurring in many or all members of one family in a pair arise from PCR errors during the first round of amplification such as might occur when copying across sites of mutagenic DNA damage. (iii) True mutations (green) present on both strands of a DNA fragment appear in all members of a family pair. Whereas artifactual mutations may co-occur in a family pair with a true mutation, all except those arising during the first round of PCR amplification can be independently identified and discounted when producing (iv) an error-corrected single-strand consensus sequence (SSCS). The sequences obtained from each of the two strands of an individual DNA duplex can then be compared to obtain (v) the duplex consensus sequence (DCS), which eliminates remaining errors that occurred during the first round of PCR.
测序完了,进行数据分析,会出现上述i、ii、iii三种情况的突变
i)在一个分子家族中只出现一次、或者少数几次的突变。而且互补分子家族没有出现同样的突变,这说明这种突变是随机错误,或者是PCR过程中后引入的复制错误,或者是Hiseq机器判读碱基有误。同时说明样本在该位置没有突变
ii)在一个分子家族中统一出现,但在与之互补的分子家族中不出现,这说明这种突变是在PCR的第一个循环中引入的复制错误。
iii)在分子家族中统一出现,而且与互补链出现对应的突变。这说明这种突变是真的、可信的。
以上中文参考陈巍学基因
论文信息:
Detection of ultra-rare mutations by next-generation sequencing
MichaelW. Schmitta, Scott R. Kennedy, Jesse J. Salk, Edward J. Fox, Joseph B. Hiatt,and Lawrence A. Loeb
Michael W. Schmitt, 14508–14513, doi: 10.1073/pnas.1208715109
文章原文:http://www.pnas.org/content/109/36/14508.full
Abstract
Next-generation DNA sequencing promises to revolutionize clinical medicine and basic research. However, while this technology has the capacity to generate hundreds of billions of nucleotides of DNA sequence in a single experiment, the error rate of ∼1% results in hundreds of millions of sequencing mistakes. These scattered errors can be tolerated in some applications but become extremely problematic when “deep sequencing” genetically heterogeneous mixtures, such as tumors or mixed microbial populations. To overcome limitations in sequencing accuracy, we have developed a method termed Duplex Sequencing. This approach greatly reduces errors by independently tagging and sequencing each of the two strands of a DNA duplex. As the two strands are complementary, true mutations are found at the same position in both strands. In contrast, PCR or sequencing errors result in mutations in only one strand and can thus be discounted as technical error. We determine that Duplex Sequencing has a theoretical background error rate of less than one artifactual mutation per billion nucleotides sequenced. In addition, we establish that detection of mutations present in only one of the two strands of duplex DNA can be used to identify sites of DNA damage. We apply the method to directly assess the frequency and pattern of random mutations in mitochondrial DNA from human cells.
1F
听师兄汇报过,确实很有想象力!