

在使用liger整合单细胞RNA-seq的文章中,我提到liger的数据结构和函数调用不及seurat那么方便和个性化,因此将两者的优势结合起来能够大大便利我们的单细胞数据分析。本文主要介绍以下两种方法:
- 使用SeuratWrappers在seurat中直接调用liger进行降维聚类
- 使用liger内置的函数在liger对象和seurat对象之间进行转换
SeuratWrappers
SeuratWrappers相当于seurat的社区工具,能够使得我们很方便地调用别的包的一些代码和方法来处理seurat对象。SeuratWrappers目前支持Monocle3、glmpca、LIGER、Harmony以及Velocity等13个常用的单细胞转录组相关的包,具体的细节可以参考GitHub。这里不再对代码做任何讲解,因为使用方法和参数与liger基本完全一致,如果有困惑可以参考使用liger整合单细胞RNA-seq。
############################################
## Project: Liger-learning
## Script Purpose: Integrating Seurat objects using LIGER
## Data: 2020.11.01
## Author: Yiming Sun
############################################
# general setting
setwd('~/sunym/project/liger_learning/')
# library
library(liger)
library(Seurat)
library(dplyr)
library(tidyverse)
library(viridis)
library(SeuratData)
library(SeuratWrappers)
# 1.Systematic comparative analysis of human PBMC
data("pbmcsca")
#pbmcsca is a seurat object
pbmcsca <- NormalizeData(pbmcsca)
pbmcsca <- FindVariableFeatures(pbmcsca,selection.method = 'vst',nfeatures = 2000)
#scale by dfferent methods --> intagrate different methods
pbmcsca <- ScaleData(pbmcsca,split.by = 'Method',do.center = FALSE)
pbmcsca <- RunOptimizeALS(pbmcsca,k = 20,lambda = 5,split.by = 'Method',max.iters = 30,thresh = 1e-06)
pbmcsca <- RunQuantileNorm(pbmcsca,split.by = 'Method',knn_k = 20,quantiles = 50,min_cells = 20,do.center = FALSE,
max_sample = 1000,refine.knn = TRUE,eps = 0.9)
#can further cluster the data and find neighbours
pbmcsca <- FindNeighbors(pbmcsca,reduction = 'iNMF',dims = 1:20)
pbmcsca <- FindClusters(pbmcsca,resolution = 0.3)
#dimension reduction and plotting
pbmcsca <- RunUMAP(pbmcsca,dims = 1:20,reduction = 'iNMF')
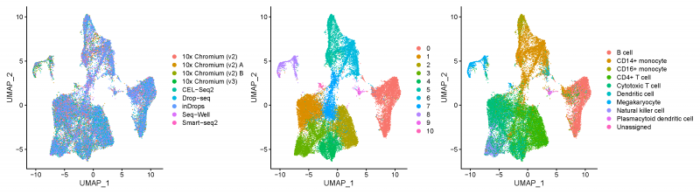
pdf(file = './res/fig_201101/pbmc_split_by_methods.pdf',width = 18,height = 5)
DimPlot(pbmcsca,group.by = c('Method','RNA_snn_res.0.3','CellType'),ncol = 3)
dev.off()
Liger内置函数
Liger包中内置了两个函数ligerToSeurat和seuratToLiger,通常我们用的比较多的是将降维聚类过后的liger对象转换成seurat对象用于做后续的差异表达分析。我们可以简单的来看一下这两个函数的效果。首先先创建一个liger对象。
############################################
## Project: Liger-learning
## Script Purpose: liger and seurat
## Data: 2020.11.14
## Author: Yiming Sun
############################################
# general setting
setwd('/data/User/sunym/project/liger_learning/')
#libarry
library(liger)
library(Seurat)
library(dplyr)
library(tidyverse)
library(viridis)
#######################################
#liger to seurat
#######################################
#load data
ctrl_dge <- readRDS("./data/PBMC_control.RDS")
stim_dge <- readRDS("./data/PBMC_interferon-stimulated.RDS")
#initialize a liger object
ifnb_liger <- createLiger(list(ctrl = ctrl_dge, stim = stim_dge))
#explore liger object
dim([email protected]$ctrl)
head(colnames([email protected]$ctrl))
head(rownames([email protected]$ctrl))
dim([email protected]$stim)
#normalize data
ifnb_liger <- normalize(ifnb_liger)
#select variable gene
ifnb_liger <- selectGenes(ifnb_liger)
#scale data but not center
ifnb_liger <- scaleNotCenter(ifnb_liger)
#integrate NMF
ifnb_liger <- optimizeALS(ifnb_liger,k = 20,lambda = 5,max.iters = 30,thresh = 1e-06)
#Quantile Normalization and Joint Clustering
ifnb_liger <- quantile_norm(ifnb_liger,knn_k = 20,quantiles = 50,min_cells = 20,do.center = FALSE,
max_sample = 1000,refine.knn = TRUE,eps = 0.9)
# you can use louvain cluster to detect and assign cluster
ifnb_liger <- louvainCluster(ifnb_liger, resolution = 0.25)
#Visualization and Downstream Analysis
ifnb_liger <- runUMAP(ifnb_liger, distance = 'cosine', n_neighbors = 30, min_dist = 0.3)
all.plots <- plotByDatasetAndCluster(ifnb_liger, axis.labels = c('UMAP 1', 'UMAP 2'), return.plots = T)
pdf(file = './res/fig_201114/plot_by_dataset_and_cluster.pdf',width = 8,height = 4)
all.plots[[1]] all.plots[[2]]
dev.off()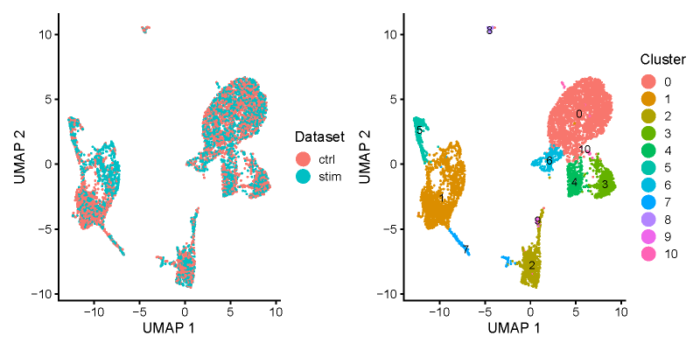
#liger to seurat #use nms ifnb_seurat <- ligerToSeurat(ifnb_liger,use.liger.genes = TRUE,by.dataset = FALSE,renormalize = TRUE) table([email protected]) head([email protected]) head(colnames(ifnb_seurat))
可以直接用ligerToSeurat函数进行转换,use.liger.genes参数表示是否保留variable gene的信息,by.dataset参数表示是否在cluster的名字之前加入dataset的名字以作区分,另外默认nms参数为names([email protected]),这个参数会在细胞的barcode之前加入dataset的名称并在orig.ident中标注出数据集的来源。可以看下输出简单理解下。
> #liger to seurat
> #use nms
> ifnb_seurat <- ligerToSeurat(ifnb_liger,use.liger.genes = TRUE,by.dataset = FALSE,renormalize = TRUE)
Warning: No assay specified, setting assay as RNA by default.
Warning: No columnames present in cell embeddings, setting to 'iNMF_1:20'
Warning: No assay specified, setting assay as RNA by default.
Warning: No columnames present in cell embeddings, setting to 'tSNE_1:2'
Warning: Feature names cannot have underscores ('_'), replacing with dashes ('-')
Performing log-normalization
0% 10 20 30 40 50 60 70 80 90 100%
[----|----|----|----|----|----|----|----|----|----|
**************************************************|
> table([email protected])
1 0 2 9 4 3 6 7 5 8 10
1500 2309 609 37 384 444 238 140 251 56 32
> head([email protected])
orig.ident nCount_RNA nFeature_RNA
ctrl_ctrlTCAGCGCTGGTCAT-1 ctrl 2232 815
ctrl_ctrlTTATGGCTTCATTC-1 ctrl 2466 760
ctrl_ctrlACCCACTGCTTAGG-1 ctrl 1085 452
ctrl_ctrlATGGGTACCCCGTT-1 ctrl 3242 925
ctrl_ctrlTGACTGGACAGTCA-1 ctrl 635 333
ctrl_ctrlGTGTAGTGGTTGTG-1 ctrl 1462 549
> head(colnames(ifnb_seurat))
[1] "ctrl_ctrlTCAGCGCTGGTCAT-1" "ctrl_ctrlTTATGGCTTCATTC-1"
[3] "ctrl_ctrlACCCACTGCTTAGG-1" "ctrl_ctrlATGGGTACCCCGTT-1"
[5] "ctrl_ctrlTGACTGGACAGTCA-1" "ctrl_ctrlGTGTAGTGGTTGTG-1"如果令nms = NULL。
> #not use nms
> ifnb_seurat <- ligerToSeurat(ifnb_liger,nms = NULL,use.liger.genes = TRUE,by.dataset = FALSE,renormalize = TRUE)
Warning: No assay specified, setting assay as RNA by default.
Warning: No columnames present in cell embeddings, setting to 'iNMF_1:20'
Warning: No assay specified, setting assay as RNA by default.
Warning: No columnames present in cell embeddings, setting to 'tSNE_1:2'
Warning: Feature names cannot have underscores ('_'), replacing with dashes ('-')
Performing log-normalization
0% 10 20 30 40 50 60 70 80 90 100%
[----|----|----|----|----|----|----|----|----|----|
**************************************************|
> table([email protected])
1 0 2 9 4 3 6 7 5 8 10
1500 2309 609 37 384 444 238 140 251 56 32
> head([email protected])
orig.ident nCount_RNA nFeature_RNA
ctrlTCAGCGCTGGTCAT-1 SeuratProject 2232 815
ctrlTTATGGCTTCATTC-1 SeuratProject 2466 760
ctrlACCCACTGCTTAGG-1 SeuratProject 1085 452
ctrlATGGGTACCCCGTT-1 SeuratProject 3242 925
ctrlTGACTGGACAGTCA-1 SeuratProject 635 333
ctrlGTGTAGTGGTTGTG-1 SeuratProject 1462 549
> head(colnames(ifnb_seurat))
[1] "ctrlTCAGCGCTGGTCAT-1" "ctrlTTATGGCTTCATTC-1"
[3] "ctrlACCCACTGCTTAGG-1" "ctrlATGGGTACCCCGTT-1"
[5] "ctrlTGACTGGACAGTCA-1" "ctrlGTGTAGTGGTTGTG-1"使用seurat的函数做个性化的差异表达分析,参考文章Seurat进行单细胞RNA-seq聚类分析。
#use liger cluster as cell type and do the DE analysis
new.cluster.ids <- c("a", "b", "c", "d", "e", "f", "g", "h", "i", "j", "k")
names(new.cluster.ids) <- levels([email protected])
ifnb_seurat <- RenameIdents(ifnb_seurat, new.cluster.ids)
ifnb_seurat$cell_type <- [email protected]
Idents(ifnb_seurat) <- 'cell_type'
all.marker <- FindAllMarkers(ifnb_seurat,only.pos = TRUE,min.pct = 0.25,logfc.threshold = 0.25)
cluster_a_vs_b_marker <- FindMarkers(ifnb_seurat,group.by = 'cell_type',ident.1 = 'a',ident.2 = 'b',only.pos = TRUE)手动导出liger中的降维图并导入seurat。
#get the tsne manually tsne.obj <- CreateDimReducObject(embeddings = [email protected],key = 'testUMAP_',global = TRUE) ifnb_seurat[['tsne']] <- tsne.obj pdf(file = './res/fig_201114/dimplot_tsetUMAP.pdf',width = 9,height = 5) DimPlot(ifnb_seurat,group.by = 'cell_type') dev.off()
注意tsne.obj的barcode要与seurat中的barcode相对应,因此可以将nms设为NULL或者为tsne.obj手动加上dataset的标签。
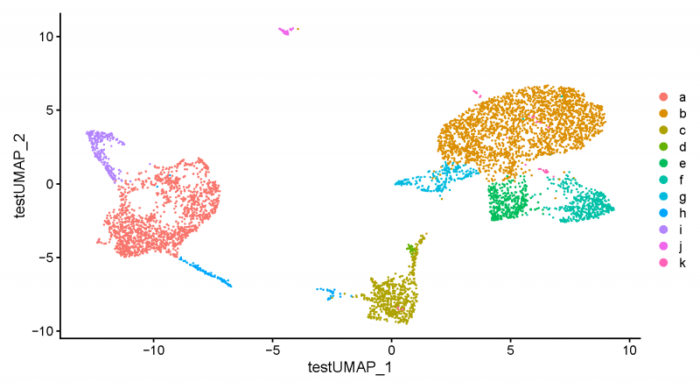
可以看到key参数中的内容被成功导入进去了。最后我们也可以将seurat对象转换为liger对象。
> ################################################### > #seurat to liger > ################################################### > ifnb_liger <- seuratToLiger(ifnb_seurat,combined.seurat = TRUE,meta.var = 'orig.ident',renormalize = TRUE) > head([email protected]) ctrlTCAGCGCTGGTCAT-1 ctrlTTATGGCTTCATTC-1 ctrlACCCACTGCTTAGG-1 a a b ctrlATGGGTACCCCGTT-1 ctrlTGACTGGACAGTCA-1 ctrlGTGTAGTGGTTGTG-1 a c b Levels: a b c d e f g h i j k > head([email protected]) tSNE_1 tSNE_2 ctrlTCAGCGCTGGTCAT-1 -9.5633178 -1.5025842 ctrlTTATGGCTTCATTC-1 -7.4026990 -0.5618219 ctrlACCCACTGCTTAGG-1 3.7179575 4.9839707 ctrlATGGGTACCCCGTT-1 -11.0730367 -4.6024990 ctrlTGACTGGACAGTCA-1 0.5273923 -8.4171533 ctrlGTGTAGTGGTTGTG-1 8.8018236 5.2826267
写在最后
这部分的内容比较枯燥,主要是我自己探索了一下seurat和liger的数据结构以及他们之间如何进行相互转换,想在这里记录一下以免自己忘了。