

目录
1. 标准化
1.1. House-keeping gene(s)
1.2. spike-in
1.3. CPM
1.4. TCS
1.5. Quantile
1.6. Median of Ration
1.7. TMM
2. 为什么说FPKM和RPKM都错了?
2.1. FPKM和RPKM分别是什么
2.2. 什么样才算好的统计量
2.3. FPKM和RPKM犯的错
2.4. TPM是一个合适的选择
1. 标准化
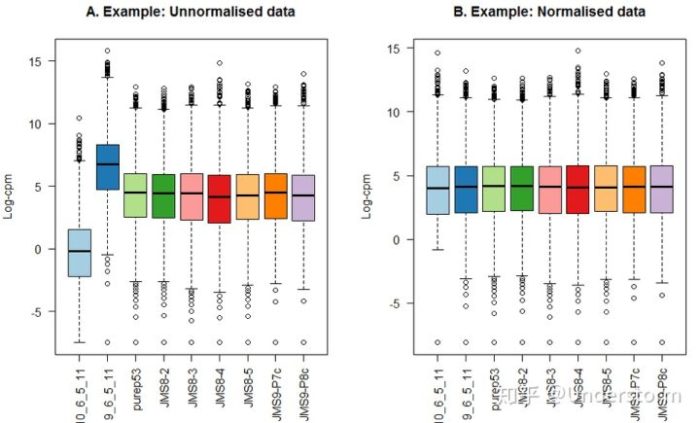
由于不同文库测序深度不同,比较前当然要进行均一化!用总reads进行均一化可能最简单,其基于以下两个基本假设:
- 绝大多数的gene表达量不变;
- 高表达量的gene表达量不发生改变;
但在转录组中,通常一小部分极高丰度基因往往会贡献很多reads,如果这些“位高权重”的基因还是差异表达的,则会影响所有其它基因分配到的reads数,而且,两个样本总mRNA量完全相同的前提假设也过于理想了。那如何比较呢,各个方家使出浑身解数,有用中位数的,有用75分位数的,有用几何平均数的,有用TMM(trimmed mean of Mvalues)的等等,总之要找一个更稳定的参考值。
1.1. House-keeping gene(s)
矫正的思路很简单,就是在变化的样本中寻找不变的量
那么在不同RNA-seq样本中,那些是不变的量呢?一个很容易想到的就是管家基因 (House-keeping gene(s))
那么 Human 常用的 House-keeping gene 怎么确定?
目前大家用的比较多的一个human housekeeping gene list 来源于下面这篇文章,是2013年发表在 Cell系列的 Trends in Genetics 部分的一篇文章

1.2. spike-in
使用Housekeeping gene的办法来进行相对定量,这种办法在一定程度上能够解决我们遇到的问题。但其实这种办法有一个非常强的先验假设:housekeeping gene的表达量不怎么发生变化。其实housekeeping gene list有几千个,这几千个基因有一定程度上的变化是有可能的
spike-in方法:在RNA-Seq建库的过程中掺入一些预先知道序列信息以及序列绝对数量的内参。这样在进行RNA-Seq测序的时候就可以通过不同样本之间内参(spike-in)的量来做一条标准曲线,就可以非常准确地对不同样本之间的表达量进行矫正
比较常用的spike-in类型:ERCC Control RNA
ERCC = External RNA Controls Consortium
ERCC就是一个专门为了定制一套spike-in RNA而成立的组织,这个组织早在2003年的时候就已经宣告成立。主要的工作就是设计了一套非常好用的spike-in RNA,方便microarray,以及RNA-Seq进行内参定量

1.3. CPM
CPM(count-per-million)
1.4. TCS (Total Count Scaling)
简单来说,就是找出多个样本中library size为中位数的样本,作为参考样本,将所有的样本的library size按比例缩放到参考样本的水平
选择一个library size为中位数的sample,以它为baseline,计算出其它sample对于baseline的normalization factor,即一个缩放因子:
然后基于该缩放因子对特定的sample中的每个基因的read count进行标准化(缩放):
1.5. Quantile
简单来说,就是排序后求平均,然后再回序
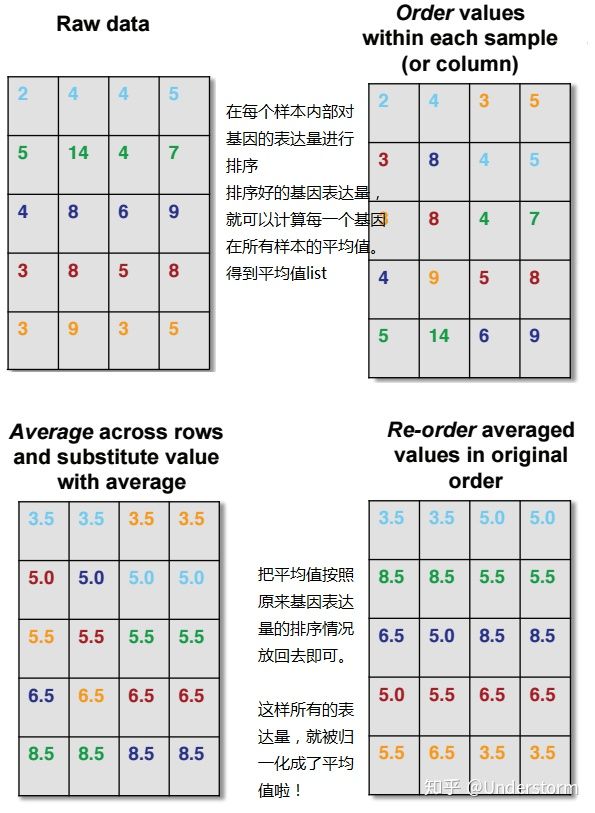
在R里面,推荐用preprocessCore 包来做quantile normalization,不需要自己造轮子啦!
但是需要明白什么时候该用quantile normalization,什么时候不应该用,就复杂很多了
1.6. Median of Ratio (DESeq2)
该方法基于的假设是,即使处在不同条件下的不同个样本,大多数基因的表达是不存在差异的,实际存在差异的基因只占很小的部分那么我们只需要将这些稳定的部分找出来,作为标准化的内参,依据内参算出各个样本的标准化因子
(1)对每个基因计算几何平均数,得到一个假设的参考样本(pseudo-reference sample)
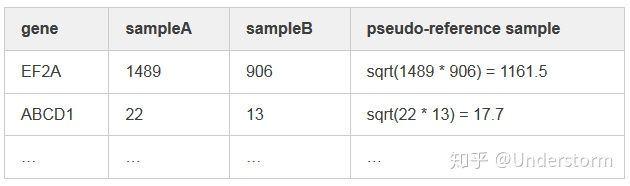
(2)对每个样本的每个基因对于参考样本计算Fold Change
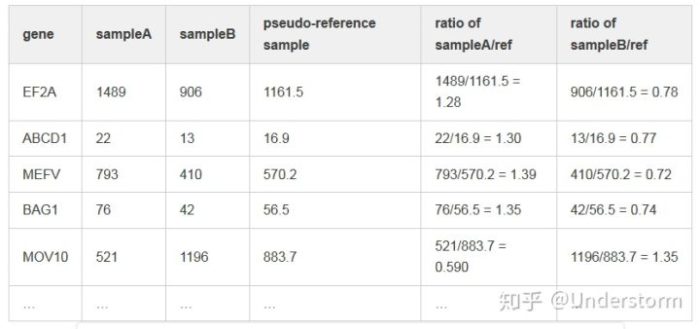
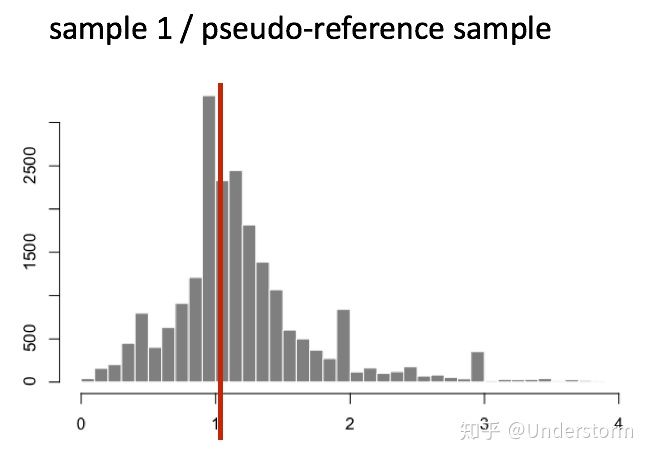
(3)获取每个样本中Fold Change的中位数,我们就得到了非DE基因代表的Fold Change,该基因就是我们选择的该样本的内参基因,它的Fold Change就是该样本的标准化因子
normalization_factor_sampleA <- median(c(1.28, 1.3, 1.39, 1.35, 0.59))
normalization_factor_sampleB <- median(c(0.78, 0.77, 0.72, 0.74, 1.35))1.7. TMM (Trimmed Mean of M value, edgeR)
该方法的思想与DESeq2的Median of Ratio相同,假设前提都是:大多数基因的表达是不存在差异的
它与DESeq2的不同之处在于对内参的选择上:
DESeq2选择一个内参基因,它的Ratio/Fold-Change就是标准化因子
edgeR选择一组内参基因集合,它们对标准化因子的计算均有贡献:加权平均
(1)移除所有未表达基因
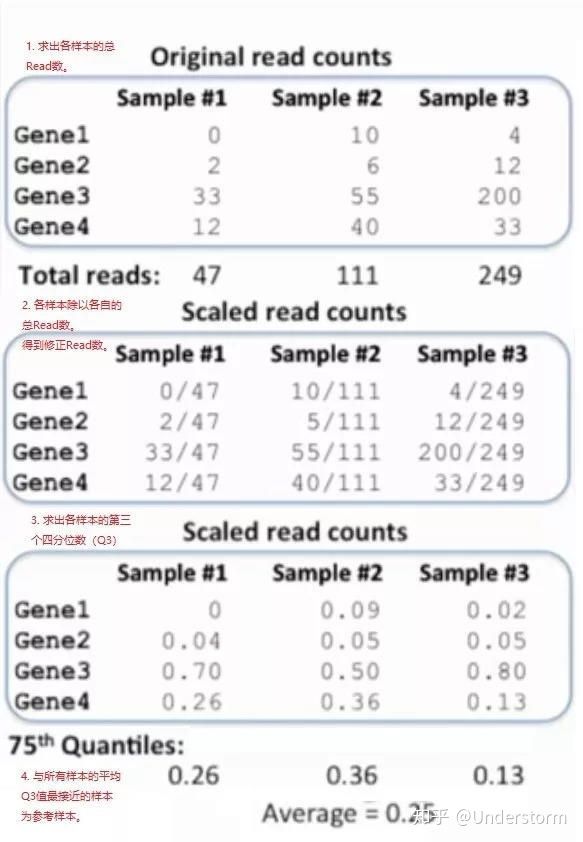
(2)从众多样本中找出一个数据趋势较为平均的样本作为参考样本
(1)对所有样本求总Read数;
(2)各样本除以各自的总Read数,得到修正Read数;
(3)求出各自样本修正Read数的Q3值(第3个四分位数);
(4)所有的Q3值求平均,与平均Q3相差最小的样本即是参考样本。
(3)找出每个样本中的代表基因集,参考这些代表基因集的fold change,计算出该样本的标准化因子
寻找样本的代表基因集:依据基因的偏倚程度和Reads数大小选出——偏倚程度小、reads数居中的基因
(1)衡量偏倚度的量:LFC (log fold change)
LFC过大或过小都表示具有偏倚性,LFC越大表示reads数在samplei中越高,即偏向samplei;LFC越小表示reads数在ref中越高,即偏向ref
(2)衡量reads数的量:read的几何平均数 (read geometric mean, RGM)
RGM越大表示基因reads越多,RGM越小表示基因reads越少
结合偏倚度、read数画出散点图:
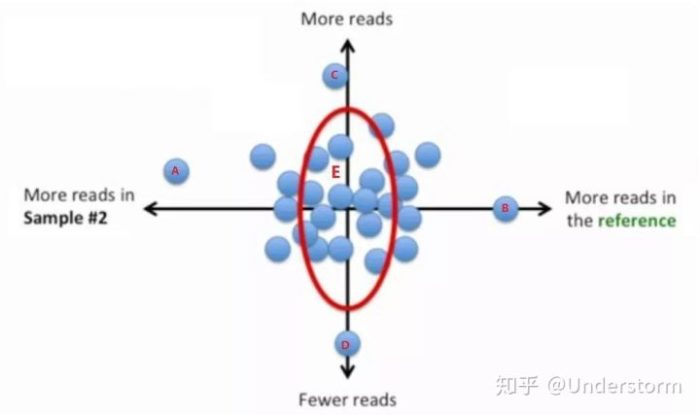
偏倚度小、表达量居中的那些基因落在图中的红线附近
由参考代表基因集计算样本的标准化因子:
对这些代表基因集计算加权平均数:
该加权平均数就能代表该样本的标准化因子,只是经过了log变换,所以需要恢复为它的正值:
2. 为什么说FPKM和RPKM都错了?
2.1. FPKM和RPKM分别是什么
FPKM和RPKM分别是什么
RPKM是Reads Per Kilobase per Million的缩写,它的计算方程非常简单:
FPKM是Fregments Per Kilobase per Million的缩写,它的计算与RPKM极为类似,如下:
与RPKM唯一的区别为:F是fragments,R是reads,如果是PE(Pair-end)测序,每个fragments会有两个reads,FPKM只计算两个reads能比对到同一个转录本的fragments数量,而RPKM计算的是可以比对到转录本的reads数量而不管PE的两个reads是否能比对到同一个转录本上。如果是SE(single-end)测序,那么FPKM和RPKM计算的结果将是一致的。
这两个量的计算方式的目的是为了解决计算RNA-seq转录本丰度时的两个bias:
- 相同表达丰度的转录本,往往会由于其基因长度上的差异,导致测序获得的Read(Fregment)数不同。总的来说,越长的转录本,测得的Read(Fregment)数越多;
- 由测序文库的不同大小而引来的差异。即同一个转录本,其测序深度越深,通过测序获得的Read(Fregment)数就越多。
2.2. 什么样才算好的统计量
首先,到底什么是RNA转录本的表达丰度这个问题
对于样本X,其有一个基因g被转录了mRNA_g次,同时样本X中所有基因的转录总次数假定是mRNA_total, 那么正确描述基因g转录丰度的值应该是:
则一个样本中基因表达丰度的均值为
而
所以
这个期望值竟然和测序状态无关!仅仅由样本中基因的总数所决定的
也就是说,对于同一个物种,不管它的样本是哪种组织(正常的或病变的),也不管有多少个不同的样本,只要它们都拥有相同数量的基因,那么它们的r_mean都将是一致的
由于上面的结果是在理论情况下推导出来的,实际上我们无法直接计算这个r,那么我们可以尝试通过其他方法来近似估计r,只要这些近似统计量可以隐式地包含这一恒等关系即可
2.3. FPKM和RPKM犯的错
实际数据来证明
假定有两个来自同一个个体不同组织的样本X和Y,这个个体只有5个基因,分别为A、B、C、D和E它们的长度分别如下:

值是:
如果FPKM或RPKM是一个合适的统计量的话,那么,样本X和Y的平均FPKM(或RPKM)应该相等。
以下这个表格列出的分别是样本X和Y在这5个基因分别比对上的fregment数和各自总的fregment数量:
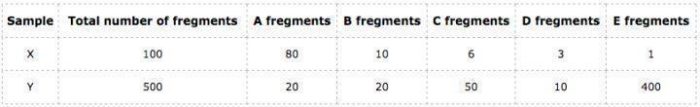
可以算出:样本X在这5个基因上的FPKM均值FPKM_mean = 5,680;样本Y在这5个基因上的FPKM均值FPKM_mean = 161,840
很明显,它们根本不同,而且差距相当大
究竟为什么会有如此之大的差异?
可以从其公式上找到答案
首先,我们可以把FPKM的计算式拆分成如下两部分。
第一部分的统计量是对一个基因转录本数量的一个等价描述(虽然严格来讲也没那么等价):
第二部分的统计量是测序获得的总有效Fregment数量的百万分之一:
这么一拆,就可以看出这个公式的问题了:逻辑上根本说不通嘛!
尤其是第二部分(
),本来式子的第一部分是为了描述一个基因的转录本数量,那么正常来讲,第二部分就应该是样本的转录本总数量(或至少是其总数量的等价描述)才能形成合理的比例关系,而且可以看出来FPKM/RPMK是有此意的,这本来就是这个统计量的目的。
但是
2.4. TPM是一个合适的选择
这个统计量在2012年所发表的一篇讨论RPKM的文章(RPKM measure is inconsistent among samples. Wagner GP, Kin K, Lynch VJ. Theory Biosci. 2012.)中就被提出来了,称之为TPM —— Transcripts Per Million,它的计算是:
简单计算之后我们就可以发现TPM的均值是一个独立于样本之外的恒定值,它等于:
这个值刚好是r_mean的一百万倍,满足等价描述的关系。