

一:下载安装该软件
下载htseq这个python模块安装解压包,依赖于很多python的其它安装包及库,模块,我最讨厌python了,在有些电脑上特别难安装,而且服务器还有权限的问题。
解压进入该目录,输入 python setup.py install –user 记住,是- – 而不是—
这样只是把这个软件安装到自己的目录
安装完毕后,会出现这两个程序,在自己的python库里面,可以直接调用这两个程序的,我这里它们的路径是 .local/bin ,很奇怪的一个路径,我也是用find命令才找到的
![]()
我在这里主要讲解,在这里调用这个命令来进行操作,直接把它当做一个程序来使用,而不是仅仅当做是python里面的一个模块调用,不需要import HTseq。
二:准备数据
输入文件
输入为sam格式的文件,如果是paired-end数据必须按照reads名称排序(sort by name)。先用samtools先对bam文件(tophat2的输出结果为bam)排序,再转换为sam。
命令:
samtools sort -n file.bam #sort bam by name samtools view -h bamfile.bam>samfile.sam
其实可以是任意的sam文件,在这里我主要演示我自己跑tophat出来的bam文件转为的sam文件,就是三个RNA数据的结果
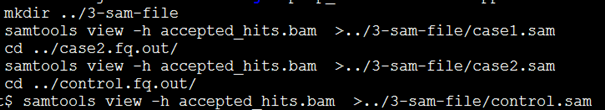
这样得到的三个sam文件特别大,bam文件是sam的二进制文件才三五个G,到了sam格式就是十几二十个G了,其实完全没必要自己把它转为sam文件,因为htseq有个参数-f可以控制输入格式是bam文件。
三:运行命令
官方的Usage:htseq-count [options]
HTSeq的作者Simon Anders建议使用ENSEMBL的gtf文件。 但是如果用了ensembl的,那么之前tophat就应该用ensembl的gtf作为参考来比对
也可以使用python -m HTSeq.scripts.count instead of htseq-count
我的命令是:
/home/jmzeng/.local/bin/htseq-count case1.sam /home/jmzeng/ref-database/hg19.gtf
但是我还是喜欢批处理来运行,一次性解决所有的bam文件计数问题
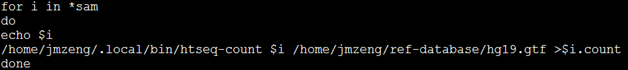
出来得到的日志是这样的
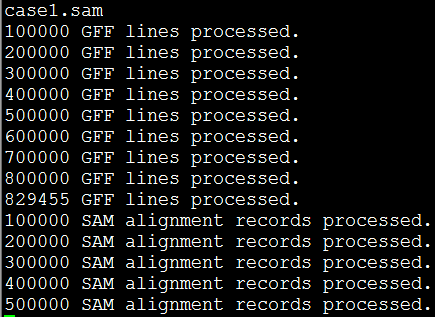
约等待几个小时就OK啦
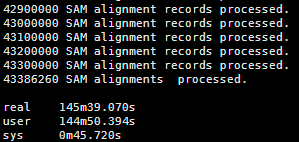
四:输出文件解读
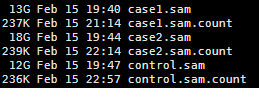
共两万多个基因,每个基因一行,基因名加上count数
![]()
可以head看一下里面的内容如下
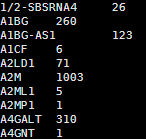
tips; 1,你可以用–idattr transcript_id来指定程序计算转录本而不是基因,但是这样会导致共有转录本重合地方太多
参考
安装:
https://www.plob.org/article/9633.html
操作htseq的方法:
http://www-huber.embl.de/users/anders/HTSeq/doc/tour.html