

写在前面
- 以下内容均来自我在菲沙基因(Frasergen)暑期生信培训班上记录的课堂笔记
1.Hi-C 辅助组装(PGA)技术原理
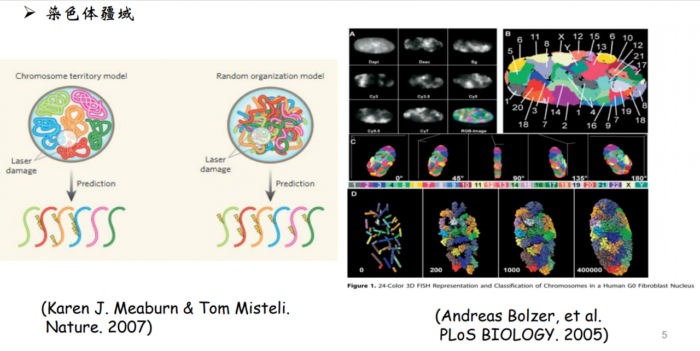
染色质在细胞核内分布的并不是随机分布的,而是不同染色体占据不同的空间。
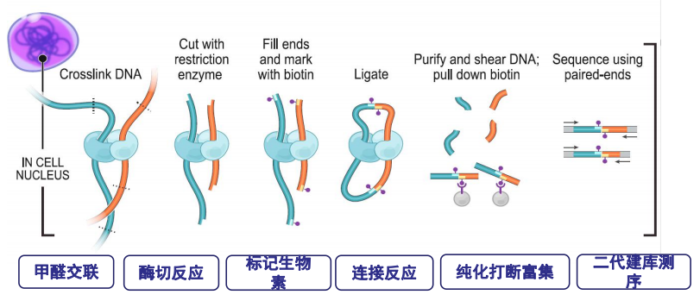
- Hi-C实验原理图
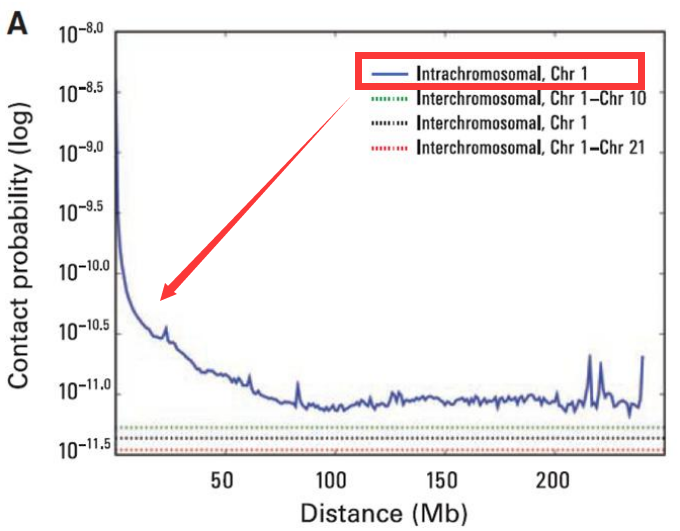
- 基因组互作衰减
染色体内互作强度较强,但也随着空间距离的增大互作强度在衰减
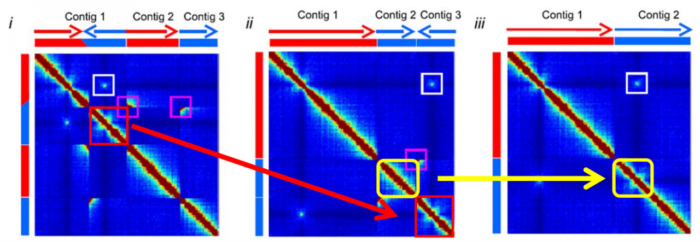
- Hi-C互作三大规律
- 1.染色体内互作富集(Intrachromosomal interaction enrichment)
- 2.互作随距离衰减(distance-dependent interaction decay)
- 3.局部互作平滑(local interaction smoothness)
2.Hi-C技术辅助基因组组装目的
- 将contigs组装到假染色体层面,实现基因组组装到染色体层面
3.Hi-C技术主流辅助组装软件
- 首选软件:Lachesis
- 辅助组装步骤
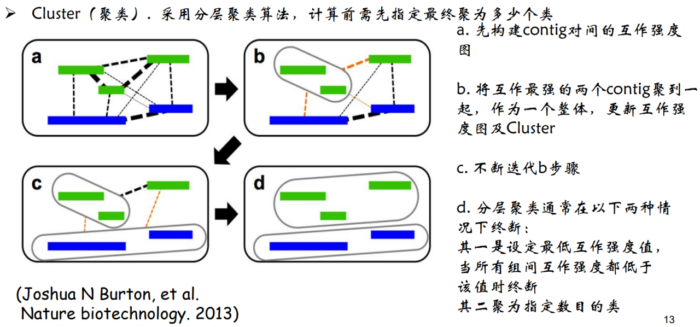
1.Cluster(聚类)。因为染色质内的互 作强度要高于染色质间的互作强度,所 以先对contig/scaffold进行聚类成染色体群。
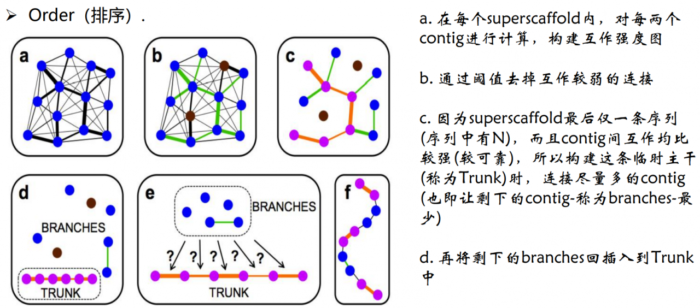
2.Order(排序)。确定每个染色体群中contig/scaffold的顺序
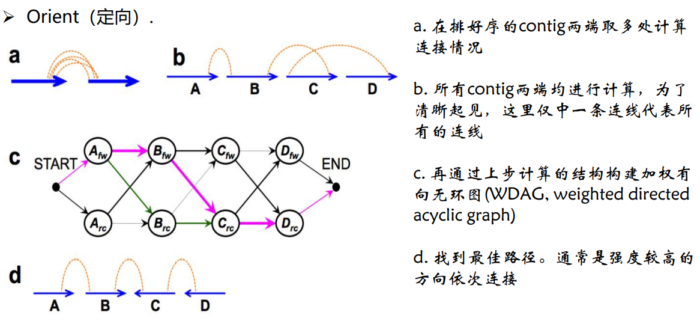
3.Orient(定向)。确定每个 contig/scaffold的方向三个步骤按照互作强度依次
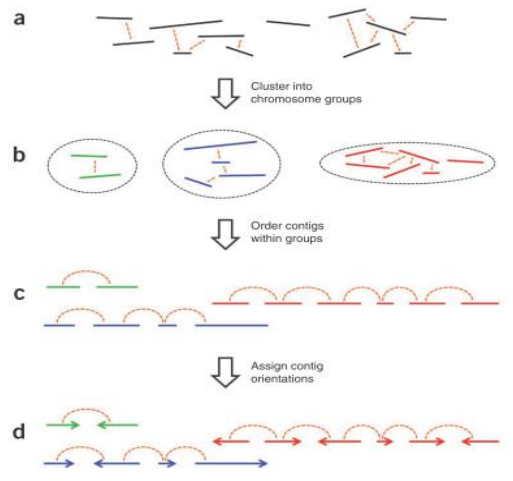
- Cluster聚类原理
- Order排序原理
- Orient定向原理
4.Hi-C PGA代码实操(PGA即辅助组装)
4.1 软件安装与编译
git clon https://github.com/shendurelab/LACHESIS.git #此外还需要使用conda安装boost与samtools conda install -c conda-forge boost-cpp -y conda install -c bioconda samtools -y conda install -c bioconda bwa -y #都安装完成后,开始编译 cd LACHESIS/ make #后续用到的脚本都在/lachesis/src/bin目录下
4.2 数据比对
- 在基因组草图目录中建立bwa索引
bwa index draft_genome.fasta
- 采用bwa-mem模式,将Hi-C reads比对到基因组草图,生成sam文件
bwa mem -t 10 ./draft_genome.fasta ./R1.fastq.gz ./R2.fastq.gz > thaliana.sam
4.3 数据过滤
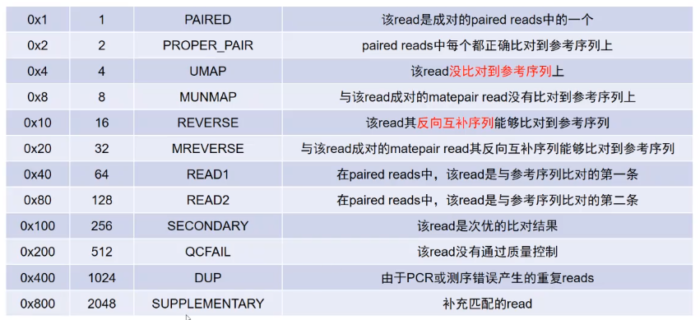
- 使用lachesis首先过滤掉flagstat大于2048的序列(flagstat > 2048表示去除补充匹配的reads见下图)
perl /lachesis/PreprocessSAMs-rmsubalig.pl thaliana.sam thaliana.II.sam #PreprocessSAMs-rmsubalig.pl脚本接输入和输出文件的名字,运行完会得到一个过滤后的sam文件:后缀.II.sam
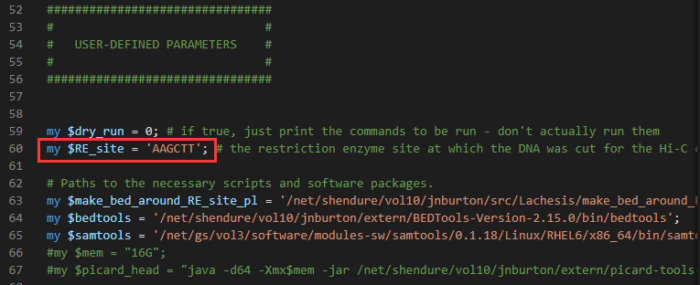
- 将PreprocessSAMs.pl程序复制到当前目录,并加以修改
cp /lachesis/src/bin/PreprocessSAMs.pl ./ vim PreprocessSAMs.pl #小tips:vim下使用set nu显示行号,输入数字直接跳转 #根据Hi-C实验过程中使用的限制性内切酶,更改酶切位点序列 (拟南芥使用的是HindIII,对应的酶切位点序列是AAGCTT,$RE_site对应的就是酶切序列)
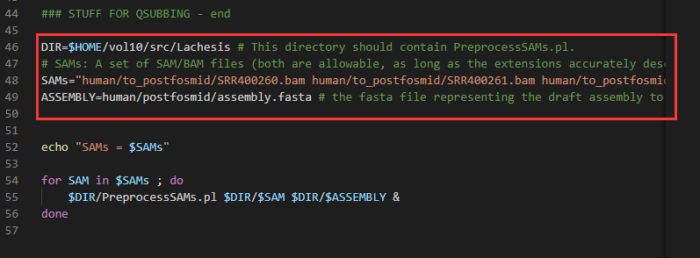
- 将PreprocessSAMs.sh程序复制到当前目录,并加以修改
cp /lachesis/src/bin/PreprocessSAMs.sh ./ vim PreprocessSAMs.sh #以下几处需修改: #DIR #在第46行,修改为目前所在的目录路径,可用pwd命令显示当前路径 #SAMS #在第48行,修改为上一步生成的sam文件名字 (例如thaliana.II.sam) #ASSEMBLY #在第49行,修改为参考基因组的路径(例如../01.ref/arabidopsis_thaliana_draft_genome.fasta) #注意:ASSEMBLY一定要用相对路径,否则会报错PreprocessSAMs.pl: Can't find draft assembly fil
- 然后过滤出距离酶切位点500bp以内且唯一比对的reads
sh PreprocessSAMs.sh # 注意:这一步运行完需要按回车键结束 cd ..
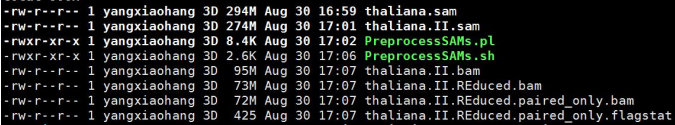
结果文件
4.4 Lachesis组装
- 建立一个lachesis分析文件夹并进入,例如:
mkdir 04.lachesis cd 04.lachesis
- 新建一个过滤文件的目录,同时建立过滤文件的软链接
mkdir bam_file cd bam_file ln -s /data/alnbam/*.II.REduced.paired_only.bam cd ..
- 复制一个lachesis的配置文件到当前目录,并修改
cp lachesis/src/bin/INIs/test_case.ini ./ vim test_case.ini
以下为输入输出文件位置参数,本例需修改:
OUTPUT_DIR:第35行,建议修改为out/NNJ_90_2_3_120_10,用文件夹名来记录重要参数值
DRAFT_ASSEMBLY_FASTA:第45行,修改为参考基因组的路径,建议设为绝对路径/local_data1/draft_genome.fasta
SAM_DIR:第49行,修改为比对文件所在的路径,即bam_file
SAM_FILES:第54行,修改为过滤得到的bam文件的名字,本例中拟南芥有两组数据:samplex.II.REduced.paired_only.bam sampley.II.REduced.paired_only.bam
RE_SITE_SEQ:第58行,因为本例中是使用的是HindIII酶,其对应的序列为AAGCTT,因此酶切序列设为AAGCTT
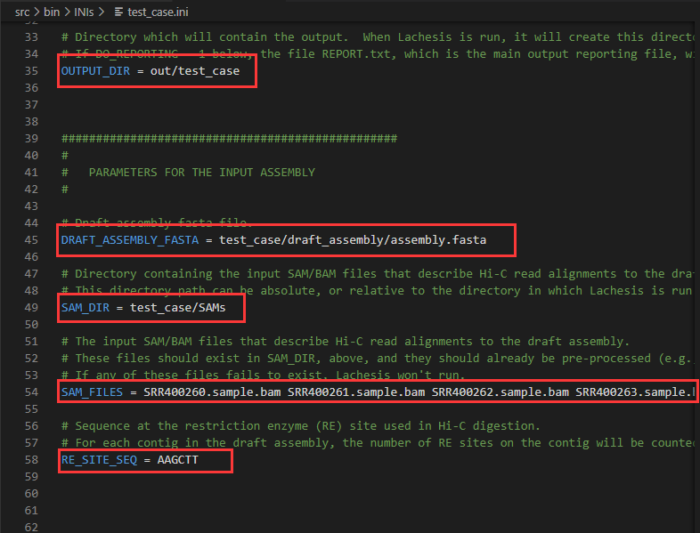
- 配置文件test_case.ini的重要参数。在实际项目中会有不同USE_REFERENCE:0代表不使用标准基因组进行评估;1表示用标准基因组进行评估(本例中不用修改)
REF_ASSEMBLY_FASTA:后面是标准参考基因组,如:REF_ASSEMBLY_FASTA = /local_data1/ninanjie.fa
BLAST_FILE_HEAD:后面加标准基因组和参考基因组的blastn比对结果,这里的格式固定,只需输入前缀BLAST_FILE_HEAD = /local_data1/ler1_vs_tari10
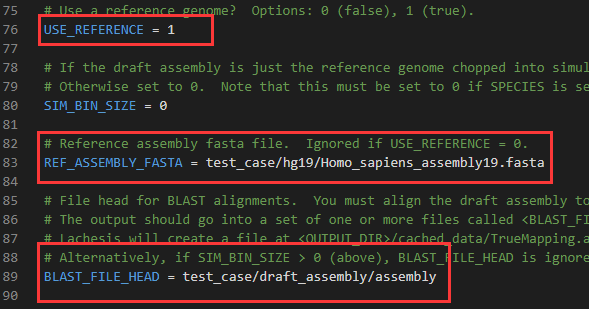
CLUSTER_N:第129行,后面接物种所对应的染色体数目,本例样本为拟南芥,因此设为5
CLUSTER_MIN_RE_SITES:第137行,聚类分析中contig/scaffold序列中的最小酶切位点数,建议设为90
CLUSTER_MAX_LINK_DENSITY:第140行,聚类分析中contig/scaffold序列中的最大Link深度,建议设为2
CLUSTER_NONINFORMATIVE_RATIO:第144行,聚类回插中contig/scaffold序列与目标cluster互作数同其他cluster互作数比例,建议设为3
ORDER_MIN_N_RES_IN_TRUNK:第153行,排序分析中TRUNK内contig/scaffold序列中的最小酶切位点数,建议设为120
ORDER_MIN_N_RES_IN_SHREDS:第155行,排序分析中回插contig/scaffold序列的最小酶切位点数,建议设为10
- 最后新建的两个目录作为组装基因组和标准基因组比对图片的存放地址
mkdir ~/bin #会在这个目录下生成画图脚本 mkdir ~/public_html #在这个目录下生成图片 ulimit -s unlimited #表示不限制线程,如果没有设置则会报错
- 运行Lachesis
/gnu_modulefile/lachesis test_case.ini
结果文件txt文件是Lachesis组装的基因和已发表的标准基因组的比较结果信息,Lachesis的组装信息在out文件夹中
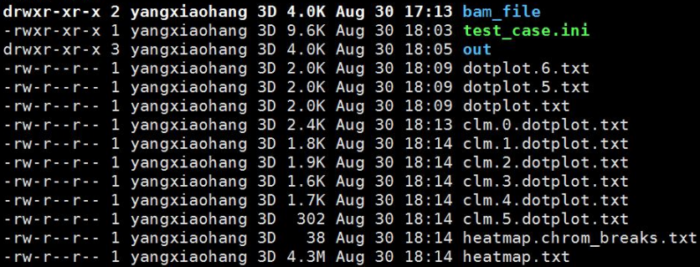
进入我们设置的out/NNJ_90_2_3_120_10
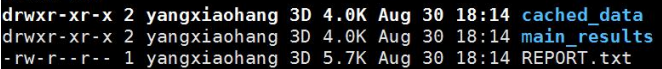
main_results/中的clusters.txt和clusters.by_name.txt展示了lachesis聚类的结果,一行代表一个group(相当于一条染色体)
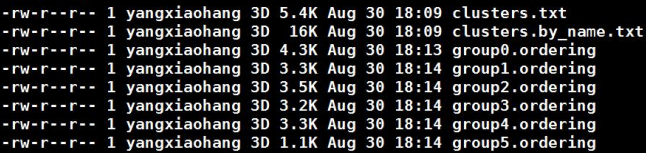
group*.ordering则展示的是每一个group中排序和方向的结果
cached_data/中则是各个group互作矩阵文件,以及在lachesis排序过程中的trunk信息。这个文件夹存放的是程序运行的中间临时文件,仅知道即可
REPORT.txt则是对Lachesis的组装结果做了一个总体的展示.REPORT.txt第一部分记录Lachesis组装全部参数;REPORT.txt第二部分记录Cluster,Order以及最后Trunk的信息:
CLUSTERING METRICS部分:如果给了近缘物种信息,会将Cluster与近缘物种进行对比,统计没有比对上的Contig数以及在染色体上位置不同的Contig数;如果项目没有提供近缘物种信息,则仅统计Cluster信息
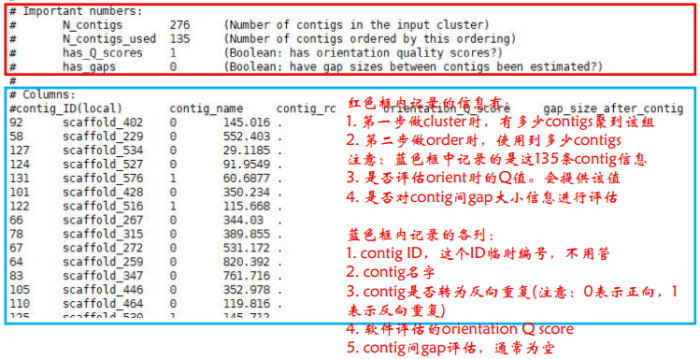
ORDERING METRICS部分:记录了orderings中contigs数目以及碱基数,trunks中的contigs数目以及碱基数。其中orderings中contigs长度与所有序列长度的比值即挂载率,如下图中红色框内的
- 通过读取main_results文件夹中的聚类、排序与定向信息,可以使用lachesis中CreateScaffoldedFasta.pl脚本生成最终的组装结果fasta文件
perl /src/bin/CreateScaffoldedFasta.pl /local_data1/draft_genome.fasta /local_data1/04.lachesis/out/NNJ_90_2_3_120_10 #输入两个参数,第一个是参考基因组路径,第二个是上一步lachesis组装时的输出文件夹路径
- 生成的最终组装文件:/local_data1//04.lachesis/out/NNJ_90_2_3_120_10/Lachesis_assembly.fasta