

ANI概念
Average Nucleotide Identity (ANI) 是在核酸水平,两两基因组之间所有直系同源蛋白编码基因的相似性,常用于研究基因组之间的进化距离。相较于传统的 DNA-DNA hybridization (DDH),ANI指标计算简单省时。且ANI指标有助于构建结构性数据库,方便生物信息学者的后续研究。还有一种计算ANI的方法是基于俩俩基因组的共同同源蛋白编码基因的相似性。接下来介绍几款计算ANI的在线工具和本地软件(Jspecies 和 pyani)。
ANI在线工具
Kostas lab:http://enve-omics.ce.gatech.edu/ani/index
MicrobeTrakr 的 ANI Calculator:http://www.microbetrakr.org/ani/
Ezbiocloud:http://www.ezbiocloud.net/tools/ani
ANI本地软件
Jspecies
http://imedea.uib-csic.es/jspecies/ DOI: 10.1073/pnas.0906412106
Jspecies是可视化操作,用户体验简单。但输入基因组相对麻烦,不方便较多基因组之间的两两计算ANI。
Jspecies安装
# install blast mkdir blast cd blast wget http://mirrors.vbi.vt.edu/mirrors/ftp.ncbi.nih.gov/blast/executables/legacy/2.2.26/blast-2.2.26-universal-macosx.tar.gz tar zxvf blast-2.2.26-universal-macosx.tar.gz -C /opt/biosoft # blast path: /opt/biosoft/blast-2.2.26/bin/blastall # formatdb path: /opt/biosoft/blast-2.2.26/bin/formatdb # fastacmd path: /opt/biosoft/blast-2.2.26/bin/fastacmd cd .. # install MUMmer mkdir MUMmer cd MUMmer wget https://sourceforge.net/projects/mummer/files/mummer/3.23/MUMmer3.23.tar.gz tar zxvf MUMmer3.23.tar.gz -C /opt/biosoft cd /opt/biosoft/MUMmer3.23 make check make install # nucmer path: /opt/biosoft/MUMmer3.23/nucmer # install Jspecies mkdir /opt/biosoft/jspecies cd /opt/biosoft/jspecies wget http://imedea.uib-csic.es/jspecies/jars/jspecies1.2.1.jar java -jar -Xms1024m -Xmx1024m jspecies1.2.jar
Jspecies运行
- 打开软件图形界面后,点击 Edit 下的 preferences,添加 blast、nucmer、formatdb、fastacmd路径;
- 设置 ANIb (基于blast得到的ANI值)
-X 空比对对应的下降值 [默认 150] -q 碱基错配罚分 [默认 -1] -F 是否过滤query序列的简单重复和低复杂度序列 [F] -e e-value [默认 1e-15] -a 运行线程数 [默认 2] Identity (%) >= 30 Alignment (%) >= 70 #more than 30% overall sequence identity (recalculated to an identity along the entire sequence) over an alignable region of at least 70% of their length length 1020 #query序列被连续切割成1020bp的片段,然后将这些片段blastn比对到reference序列,比对的结果用于后续计算
- ANIm (基于MUMmer得到的ANI值)参数
-b 低得分区大于此值时,停止比对 [默认 200] -c 一簇匹配的序列最少需要有大于此长度的匹配。此值越大,则小的匹配越少 [默认 65] -g 在一簇匹配的序列中,两个邻近的matches之间的gap的最大阈值。此值越大,则越容易找到更大匹配区 [默认 90] -l single match的最小长度 [默认 20]
- 导入数据并运行:File -> New File -> Import FASTA(S) from files -> 选择 Tetra、ANIb、ANIm -> Start
ANI命令行软件
pyani
pyani是用于计算ANI的python3模块,其依赖包有numpy、scipy、matplotlib、biopython、pandas、seaborn。0.1.3.2及以上版本的其他依赖包不用另外安装,0.1.3.2以下版本 pip install -r requirements.txt 安装依赖包。pyani 包括average_nucleotide_identity.py (计算ANI) 和 genbank_get_genomes_by_taxon.py (从NCBI下载数据),并提供四种不同算法:
- ANIm:使用 MUMmer (NUCmer) 比对序列
- ANIb:使用 BLASTN+ to 比对序列的 1020nt 片段
- ANIblastall:使用老版的 BLASTN 比对序列的 1020nt 片段
- TETRA:计算每个序列的四核苷酸的频率
pyani安装
# pip3 install pip3 install pyani echo "PATH=/home/linuxbrew/.linuxbrew/bin:$PATH" >> ~/.bashrc source ~/.bashrc # local install git clone git@github.com:widdowquinn/pyani.git cd pyani python3 setup.py install
pyani运行
# example average_nucleotide_identity.py -i tests/test_ani_data/ -o tests/test_ANIm_output -m ANIm -g average_nucleotide_identity.py -i tests/test_ani_data/ -o tests/test_ANIb_output -m ANIb -g average_nucleotide_identity.py -i tests/test_ani_data/ -o tests/test_ANIblastall_output -m ANIblastall -g average_nucleotide_identity.py -i tests/test_ani_data/ -o tests/test_TETRA_output -m TETRA -g
pyani参数
# required
-i INDIRNAME, --indir INDIRNAME 输入文件所在目录
-o OUTDIRNAME, --outdir OUTDIRNAME 输出结果目录
# optional
-v, --verbose 输出详细结果
-f, --force 强制文件的写入
-s FRAGSIZE, --fragsize FRAGSIZE
ANIb比对时序列的片段大小 [默认 1020]
-l LOGFILE, --logfile LOGFILE
输出日志文件
--skip_nucmer 跳过运行NUCmer
--skip_blastn 跳过运行BLASTN
--noclobber 不重做已存在文件
--nocompress 不压缩或删除比较结果
-g, --graphics 生成ANI的热图
--gformat GFORMAT 图形文件输出格式 [pdf|png|jpg|svg] [默认 pdf,png,eps]
--gmethod {mpl,seaborn} 图形输出方法 [默认 mpl]
--labels LABELS 序列标记文件的路径
--classes CLASSES 序列分类文件的路径
-m {ANIm,ANIb,ANIblastall,TETRA}, --method {ANIm,ANIb,ANIblastall,TETRA}
ANI比对方法 [默认 ANIm]
--scheduler {multiprocessing,SGE}
Job scheduler [默认 multiprocessing]
--workers WORKERS 运行线程数 [默认 全部线程]
--SGEgroupsize SGEGROUPSIZE
SGE组中job数 [默认 10000]
--SGEargs SGEARGS Additional arguments for qsub
--maxmatch Override MUMmer to allow all NUCmer matches
--nucmer_exe NUCMER_EXE
NUCmer 程序路径
--filter_exe FILTER_EXE
delta-filter 程序路径
--blastn_exe BLASTN_EXE
BLASTN+ 程序路径
--makeblastdb_exe MAKEBLASTDB_EXE
BLAST+ makeblastdb 程序路径
--blastall_exe BLASTALL_EXE
BLASTALL 程序路径
--formatdb_exe FORMATDB_EXE
BLAST formatdb 程序路径
--write_excel 输出Excel格式文件
--rerender Rerender graphics output without recalculation
--subsample SUBSAMPLE
Subsample a percentage [0-1] or specific number (1-n)
of input sequences
--seed SEED Set random seed for reproducible subsampling.
--jobprefix JOBPREFIX
Prefix for SGE jobs (default ANI).
pyani自带绘图,若不满意,可以调用ggplot2绘制热图
Rscript heatmap_ANI.R $OUTPUT/ANIb_percentage_identity.tab
#!/usr/bin/env Rscript
library("ggplot2")
library("reshape")
userprefs <- commandArgs(trailingOnly = TRUE)
file_name <- userprefs[1]
# read file
data.ANI <- read.table(file_name)
# cluster and order matrix
row.order <- hclust(dist(data.ANI))$order # clustering
col.order <- hclust(dist(t(data.ANI)))$order
dat_new <- data.ANI[row.order, col.order] # re-order matrix accoring to clustering
# melt to dataframe
df_molten_dat <- melt(as.matrix(dat_new)) # reshape into dataframe
names(df_molten_dat)[c(1:3)] <- c("shewanella1", "shewanella2", "ANI")
# make plot
p1 <- ggplot(data = df_molten_dat,
aes(x = shewanella1, y = shewanella2, fill = ANI)) +
geom_raster() +
scale_fill_distiller(palette = "RdYlBu", limits=c(0.8, 1)) +
theme(axis.text.x = element_text(angle = 90, hjust = 1)) +
ggtitle("Shewanella ANI heatmap")+
geom_text(aes(label = round(ANI, 2)))+
theme(axis.title=element_text(size=16), strip.text.x=element_text(size=16),
legend.title=element_text(size=15), legend.text=element_text(size=14),
axis.text = element_text(size=14), title=element_text(size=20),
strip.background=element_rect(fill=adjustcolor("lightgray",0.2))
)
# export plot
png("Heatmap_ANI_highres.png", width=15, height=15, units="in", pointsize=12, res=500)
p1
dev.off()
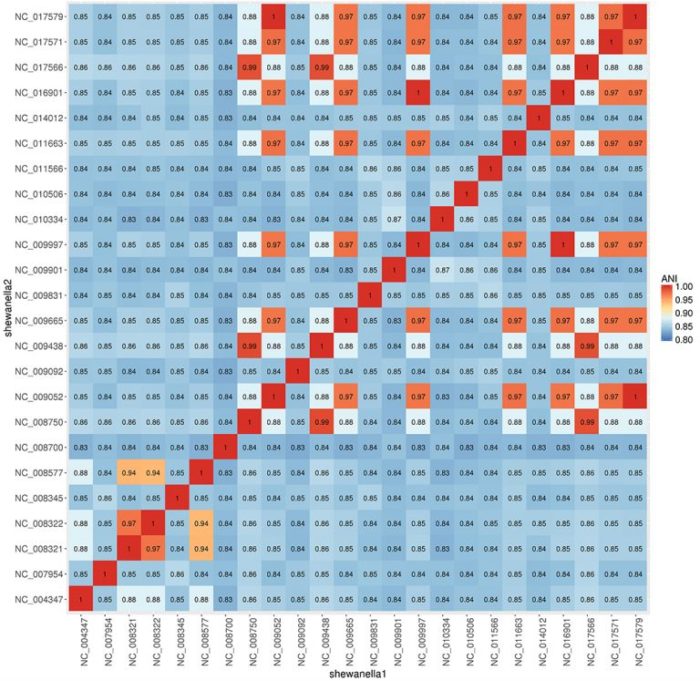
示例图效果
24种不同 shewanella 属菌种ANI比较热图
参考:
http://imedea.uib-csic.es/jspecies/
https://github.com/widdowquinn/pyani/
http://www.a-site.cn/article/300418.html
https://github.com/rprops/MetaG_lakeMI/wiki/
1F
写得好棒👍