

这次学习的本体是来自剑桥大学: Vladimir Kiselev, Tallulah Andrews, Davis McCarthy and Martin Hemberg几位大佬建立的course→Analysis of single cell RNA-seq data以及来自哈佛大学医学院的 McCarroll Lab的Computational resources的CookBook

NOTIFICATION!:10月30号和11月1号有了重大更新,目前仅介绍最新的background,方法学等更新和学习后再论述
取得翻译许可
Sure, feel free to translate and adapt the course to your audience. It is under GPL-3 license.
——Vladimir Kiselev
第二代测序技术的发展给生物学的研究带来了极大突破。高通量测序现已被用于研究的各个领域,Bulk RNA-seq作为其中的突出代表,在比较转录组学,疾病研究中发挥着极大的作用。然则其功能依然有着相对薄弱的地方,例如表达水平是一群细胞的相对平均水平,对于复杂的表达时刻变化的系统无法使用,对于基因表达的特性也无法研究。
所以single cell RNA-seq的技术也应运而生,这种技术首先由M Azim Surani及汤富酬创建于2009年,发表于
NATURE METHOD:
Tang, Fuchou, Catalin Barbacioru, Yangzhou Wang, Ellen Nordman, Clarence Lee, Nanlan Xu, Xiaohui Wang, et al. 2009. “mRNA-Seq Whole-Transcriptome Analysis of a Single Cell.” Nat. Methods 6 (5): 377–82.
但是直到14年随着方法的成熟与测序成本的降低这种方法才渐渐的进入大家的视野。
Single-cell 工作流程 OVERVIEW
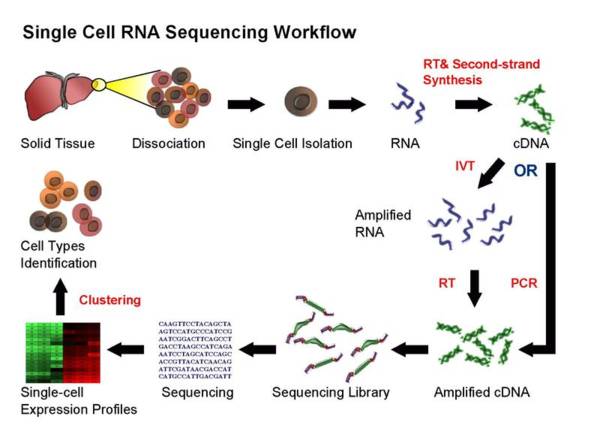
single cell sequencing (taken from Wikipedia)
以Nanoliter Droplets方法为例:
Highly Parallel Genome-wide Expression Profiling of Individual Cells Using Nanoliter Droplets
overview
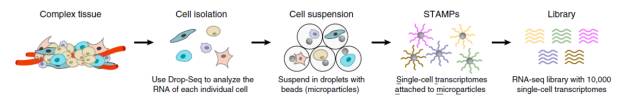
首先是组织处理得到单细胞,包裹在单个的microparticle里面,而microparticle里面又存有包含polyT的beads,于是可以结合mRNA反转成为cDNA,建成pool进行PCR扩增,最后混合所有的STAMPs高通量测序得到数据。
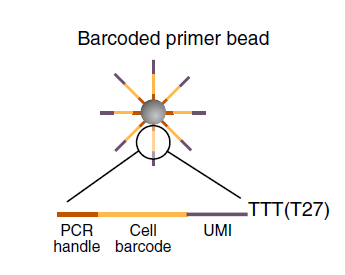
每个micro particle上面的序列由四个部分组成:
- 一段一样的序列,PCR handle用于后续的PCR扩增
- bead特异性的barcode,10 - 12bp,用来区分单个细胞,理论上存在4^12 (16,777,216)个barcode,也就是说最多可以处理1600W个细胞
- UMI,Unique Molecular Identifier,4 - 8bp,每个beads上理论存在4^8 (65,536)个UMI,用来区分transcripts,理论上可以区分6W个转录本
- 30bp的oligo-dT,用来捕捉mRNA完成反转录
这个课程呢,主要关注scRNA-seq的到的数据处理,mark黄色的部分呢,是适用于高通量测序的数据处理流程;mark橙色的部分呢,则是需要利用已有的处理RNA-seq的工具和一些新开发的区分scRNA-seq的方法;mark蓝色的部分,就是需要专门的处理scRNA-seq的软件来探究这里面的生物学意义了。
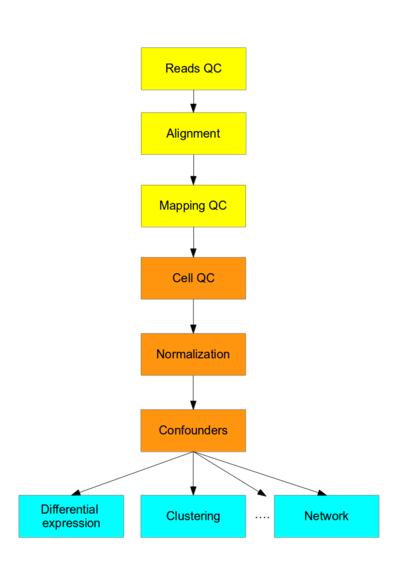
Flowchart of the scRNA-seq analysis
单细胞RNA测序区别于混池RNA测序的地方在于每个测序库(sequencing library)都代表着一个单细胞,所以我们应该将着眼点放在比较单个不同细胞上(或细胞群),这种测序库(sequencing library)的差异来源于一下两个方面:
- Amplification:扩增偏差,单个细胞初始转录本的捕捉效率和低输入会导致这样的偏差
- Gene ‘dropouts’ :基因丢失,有些基因会在某个细胞里检测到具有中等表达水平却在其它细胞里面没有被发现
以上两点也正式目前研究较多的领域,大家都致力于消除这些偏差使得数据更具有可分析性
Overview of experimental methods for generating scRNA-seq data
目前单细胞测序领域非常火热,近些年来涌现出很多测序方法,包括但不限于:
- CEL-seq
- CEL-seq2
- Drop-seq (原理介绍使用的方法)
- InDrop-seq
- MARS-seq
- SCRB-seq
- Seq-well
- Smart-seq
- Smart-seq2
- SMARTer
- STRT-seq
但是即使测序办法繁多丰富,但根底里是需要面对两个问题: quantification(定量)和 capture(捕捉)
Quantification(定量)
关于quantification(定量),目前存有着两种处理方式:full-length(全长) and tag-based(标签依赖)
full-length的处理方法旨在对每个转录本获取统一的测序覆盖度,相反tag-based处理方法只捕捉mRNA的5'或3'端,定量处理方法的选择取决于你后期想要分析的目的。
理论上,full-length的处理可以提供一个相对平均的测序覆盖度,但是就目前的结果来说还是存在着很多bias。
而tag-based的优势在于它可以结合UMI(前面介绍过)来提高定量的水平,缺点在于未捕捉完全的转录本序列,在比对的时候无法区分iosform (Archer et al. 2016)
Capture(捕捉)
捕捉RNA的策略决定了你的产出,细胞如何被选择包括是否携带额外信息都值得大做文章。三个被广泛运用的方法包括:
- microwell-based
- microfluidic-based
- droplet-based
- microwell-based
简单来说,这种方法就是把单个细胞利用laser capture或者example pipette的技术分离到微流体孔里面。这种技术的既有优势在于可以结合FACS分选技术,根据细胞表面marker挑选出的合适的细胞亚群,并且可以对细胞形态进行记录,找出并丢弃损伤细胞或粘连的非单个细胞。这个技术的缺陷在于由于分选的局限性导致的低通量,和相匹配的较大的工作量。

Image of microwell plates (image taken from Wikipedia)
microfluidic-based
以Fluidigm’s C1举例,其提供了一个整合的细胞捕获系统,并可以执行建库前的反应,所以相对于microwell-based方法有更高的通量。
但其弊端在于其只能捕获10%左右的细胞,所以不太适合应用于有较少样品量或者样品较为珍惜的情况。

Image of a 96-well Fluidigm C1 chip (image taken from Fluidigm)
droplet-based
这种技术就是我以上介绍的原理的例子,通过纳升级别的携带beads的小液滴,捕获单个细胞,并在液滴内完成建库,其优势在于可以定量的鉴别每个cell内的转录本数量,劣势在于测序深度低,往往一个细胞只有小几千个转录本被检测到。
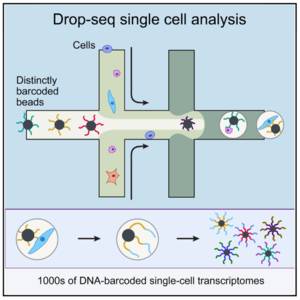
Schematic overview of the drop-seq method (Image taken from Macosko et al)
老生常谈
适合的即是最好的。
你所选用的决定于你想要研究什么样的生物学问题。
假如想要定义一个组织内细胞的组成成分,那么droplet-based方法是较为有效的,因为它可以捕捉到相对大数量级的细胞。
换一个方面来说,加入想要去研究一群数量有限而又知道细胞表面分子marker的细胞,那么FACS分选接测序才是较好的方案。
想要研究可变剪切转录本,自然需要全长测序的实验方案,这个时候tag标签和UMI就成了异常鸡肋的存在(当然在定量中还是举足轻重的,要看如何取舍了)。
Enard团队(Ziegenhain et al. 2017) 和Teichmann团队 (Svensson et al. 2017) 的通过对同一个样本(mESCs)的测序和分析比较了现存于市场上的几种测序方法,在控制细胞输入数量和测序深度的时候,作者得以探究的不同实验方案特有的敏感性/噪音水平/花费,结论可见下图:
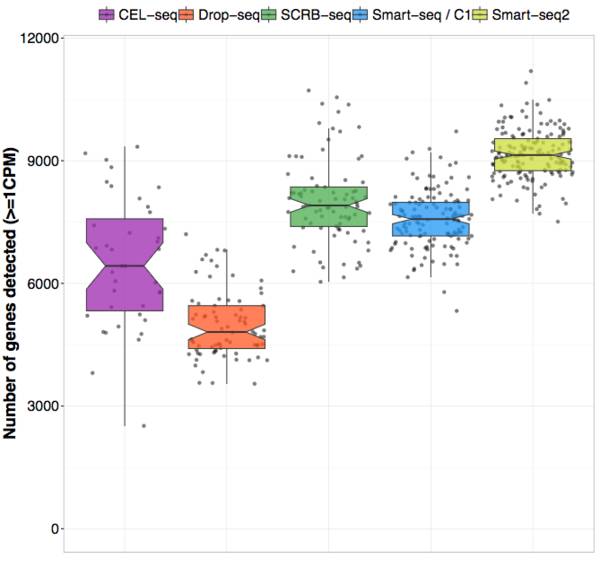
可以发现,不同的实验方案至多会造成两倍的实验差距,实验方案的抉择从一开始就决定了你的实验结果的好坏。
针对检测准确性和敏感性的探究则使用了人工合成的已知浓度的spike-in来进行。
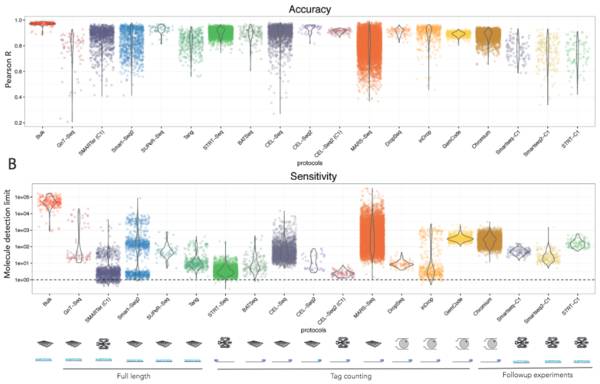
当然啦,随着时间的进展,实验方法的提高和数据分析手段的开发,我们对于这些单细胞测序方法会有更深层次的了解,但就目前而言,这个研究可以很好的帮助科研工作者选择他适合的工具并完成他的目的。