

细胞核内的染色质构像一般分以下几个层级
- Chromosome Territories
- Compartment A/B
- Topologically Associating Domains (TADs)
- Local Chromatin Interactions (Loop)
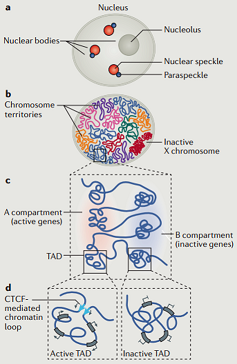
Engreitz J M. Nature Reviews Molecular Cell Biology, 2016
不同层级的染色质构像特征需要由不同分辨率的Hi-C交互矩阵进行对应的算法分析得到。 一般情况下不同层级的染色质构像特征对应的分析分辨如下:
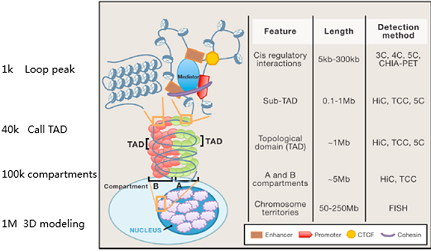
Rivera C M et al. Cell, 2013
分辨率越高对应的测序量要求越大,例如第一篇提出了Compartment概念的文章Lieberman-Aiden E et al. science, 2009,第一篇提出TAD概念的文章Dixon J R et al. Nature, 2012,测序数据量如下:
| Organism Name | Total Size, Mb | Total Spots | Total Bases | Library Name |
|---|---|---|---|---|
| Homo sapiens | 1,613 | 18,145,940 | 2,758,182,880 | Hi-C (HindIII,K562,1) 8 |
| Homo sapiens | 1,929 | 18,677,569 | 2,838,990,488 | Hi-C (HindIII,K562,1) 7 |
| Homo sapiens | 1,586 | 15,132,243 | 2,300,100,936 | Hi-C (NcoI,GM,1) 2 |
| Homo sapiens | 1,406 | 13,527,036 | 2,056,109,472 | Hi-C (NcoI,GM,1) 1 |
| Homo sapiens | 869 | 8,607,443 | 1,308,331,336 | Hi-C (HindIII,GM,2) 3 |
| Homo sapiens | 783 | 7,889,352 | 1,199,181,504 | Hi-C (HindIII,GM,2) 2 |
| Homo sapiens | 713 | 7,068,675 | 1,074,438,600 | Hi-C (HindIII,GM,Biological Repeat) |
| Homo sapiens | 622 | 6,443,641 | 979,433,432 | Hi-C (HindIII,GM,1) |
| Homo sapiens | 9,939 | 259,123,992 | 18,656,927,424 | GSM892307: RenLab-HiC-IMR90, replicate two |
| Homo sapiens | 21,572 | 496,522,946 | 35,749,652,112 | GSM892306: RenLab-HiC-hESC, replicate two |
| Mus musculus | 12,430 | 294,763,499 | 21,222,971,928 | GSM892305: RenLab-HiC-cortex, replicate two |
| Mus musculus | 16,295 | 401,291,092 | 28,892,958,624 | GSM892304: RenLab-HiC-cortex, replicate one |
| Homo sapiens | 16,479 | 397,194,480 | 28,598,002,560 | GSM862724: RenLab-HiC-IMR90 |
| Homo sapiens | 11,737 | 237,662,270 | 23,702,833,392 | GSM862723: RenLab-HiC-hESC |
| Mus musculus | 18,029 | 425,506,797 | 30,636,489,384 | GSM862722: RenLab-HiC-mESC-NCoI |
| Mus musculus | 13,243 | 340,651,343 | 24,526,896,696 | GSM862721: RenLab-HiC-mESC-HindIII, replicate two |
| Mus musculus | 26,828 | 465,473,330 | 42,892,374,448 | GSM862720: RenLab-HiC-mESC-HindIII, replicate one |
第一篇分辨率测到kb级别得文章Rao S S P et al. Cell, 2014,为了达到kb别分辨率,对GM12878 B-lymphoblastoid细胞单个样本测序量达4.9 billion pairwise reads。随着要达到的Hi-C交互矩阵分辨率的提升,需要的测序量是以指数形式递增的。如果研究者的需求只要达到TAD级别就够的话,Hi-C交互矩阵能够达到40k的分辨率就行了没有必要达到更高的分辨率,但是对于特定分辨率的Hi-C需求,怎样合理预估测序量呢?
要做到合理预估测序量我们需要对分辨率的概念进行明确。
分辨率概念
有两篇三维基因组内奠基性的文章分别给出了关于分辨率的定义:
第一种定义
Belton J M et al. Methods, 2012这篇文章第一次提出了分辨率明确定义:
对于一定测序量的Hi-C数据,把最终质控得到的unique valid pair reads map 到不同的分辨率矩阵,如果矩阵80%以上的bin pair至少有1对交互reads,那么就说这些测序数据达到了这个矩阵的分辨率。
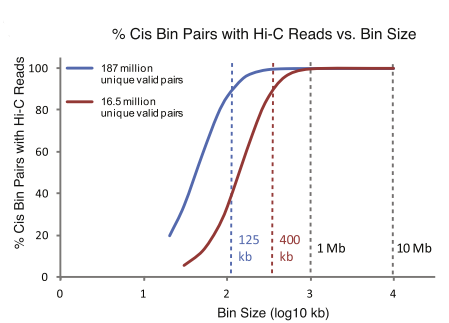
第二种定义
Rao S S P et al. Cell, 2014这篇文章达到了950bp的分辨率,它的定义是这样的:
对于一定测序量的Hi-C数据来说,对基因组按特定的窗口(bin)大小进行划分(例如1kb),如果最终80%以上的bin都至少有1000条unique valid reads覆盖,那么就说这些测序数据达到了这个bin size的分辨率。
我们可以把特定的测序数据做成不同分辨率的矩阵,矩阵分辨率越高,相应的矩阵也就越稀疏(不饱和),以上两种定义本质上其实都是一样的,都是在以特定数据达到的矩阵的饱和度进行定义,第一种定义更加直接,第二种定义在操作上更好计算一些,类似于测序深度的概念,这个测序深度不同于重测序中的单碱基的测序深度,而是特定窗口(bin)的测序深度(落在bin里的测序reads数)。
我们预测特定分辨率的Hi-C数据测序量以上述第二种分辨率的定义为基础,但是要对具体参数稍微做一下修改:80%以上的bin至少覆盖度的unique valid reads数
1000需要做一些讨论和修改。
80%以上的bin至少覆盖度的unique valid reads达到多少合适?
文章给出的答案是1000,但是这个1000具体是怎么来的呢?这个1000真的合适吗?
一个可接受的Hi-C交互矩阵热图一般是这样的:
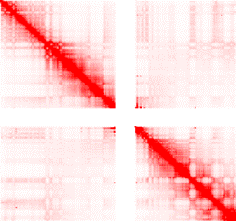
这个矩阵的主要特点是距离近的基因组位点之间的交互数值一般比距离远的位点间的交互数值要高(测得的Hi-C unique valid reads多),一般由IDEs(Interaction decay exponents)来描述这种随距离的增大交互数值衰减的程度。
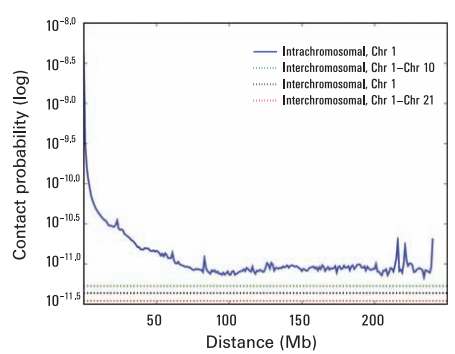
Lieberman-Aiden E et al. science, 2009
大多数物种的Hi-C交互矩阵的IDE都和距离幂次线性相关,如下图:
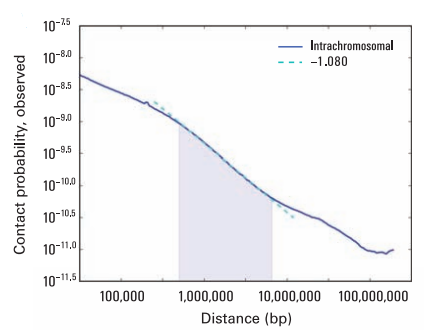
那么对于某一个特定的bin X来说,与X不同距离的各bin与X的交互也会遵循这个IDE的规律,要满足这个规律那么X所测得的unique valid reads(深度)至少要有一个最低值(通过IDE的斜率 截距累积计算)。根据我们的经验不同物种不同分辨率矩阵的这个IDE的斜率多少有些差异,算出来的X的最低深度大概在1000-4000之间比较合适。
影响预测数据量的数据质控参数
前面我们确定了80%以上的bin至少覆盖度unique valid reads在1000-4000之间比较合适,但是bin的总数以及raw reads到unique valid reads的数据质控损耗比例就需要确定了。
bin的总数可以由基因组大小除以要达到的分辨率大小确定。影响unique valid reads最终有效比例的因素包括:
- Celan Reads的比例
- 比对到基因组的Unique Mapped Ratio
- Interaction Rate
- Interaction Reads的Pcr dup Ratio
- Cis Ratio
结合以上各因素的经验值我们就能预测最终需要测序的Raw data多少了,还可以根据每个Hi-C文库一般的数据产出确定要建几个Hi-C文库。