DNAstar软件共有七个小程序(见下图),各自执行不同的功能,Editseq用于序列编辑,Seqman可以去除载体序列和拼接序列,MegAlign则主要执行序列比对的功能.
开启MegAlign软件首先需要点击File---New 新建一个工作文件
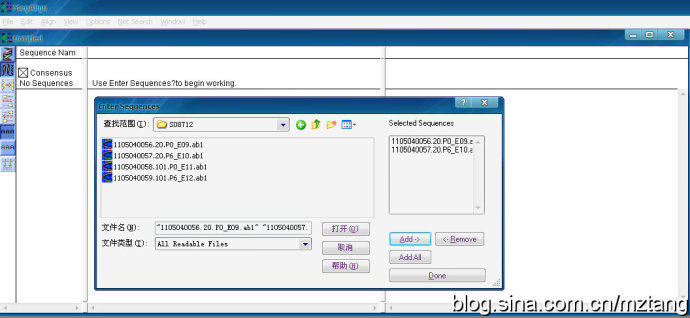
再点击File---Entersequeces 添加需要比对的序列,支持多种格式的文件(.seq;.abi;.pro;.fas等),点Add可添加多个文件,点Done导入选中的文件.
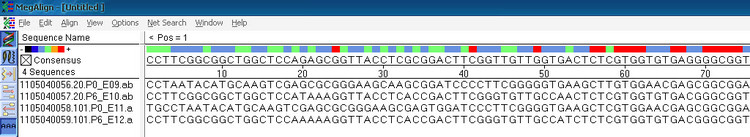
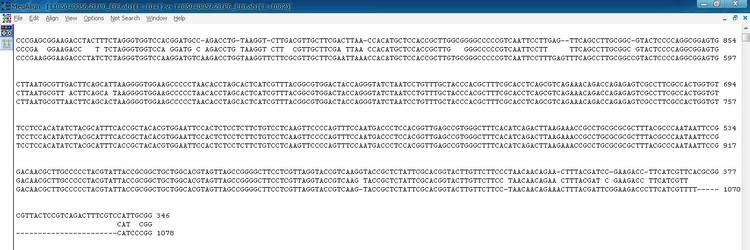
这是文件导入后的界面
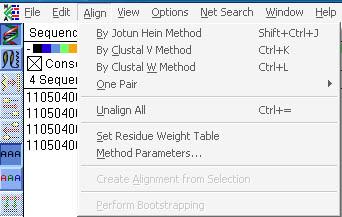
点Align选择序列比对的算法,其中多序列比对有三种算法:The Jotun Hein method,ClustalV和Clustal W;一般选择Clustal W即可。三种算法的差异如下:
“The Jotun Hein method was devised to align sequences that are previously known to be related by descent. The ClustalV and ClustalW alignment methods do not make this assumption.
ClustalW is an advancement over ClustalV, and was designed to create more accurate alignments for highly diverged sequences”。
“CLUSTALW 是一种渐进的多序列比对方法,先将多个序列两两比对构建距离矩阵,反应序列之间两两关系;然后根据距离矩阵计算产生系统进化指导树, 对关系密切的序列进行加权;然后从最紧密的两条序列开始,逐步引入临近的序列并不断重新构建比对,直到所有序列都被加入为止”。
红色区域表示的是相似性最高的区域,蓝色和绿色是同源性较低的区域;
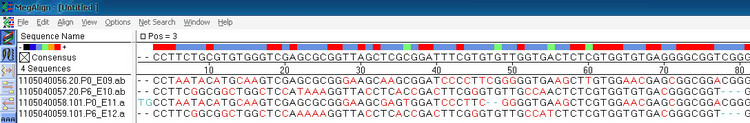
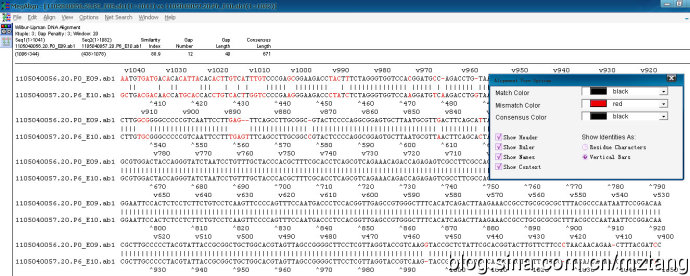
多序列比对的一个缺点就是不会自动对序列反向互补后再比对,所以反向互补后同源性高的序列检测不出来。如下图中的20.p0和20.p6其实是同一模板的两个方向的测序结果,像这种需要用到两两比对(One pair)。
One Pair也有好几种算法,任选一种都可,有兴趣的可以自己查找一下差别;
One Pair比对时需要先选中将要比对的两个序列
这是比对后的默认结果,不太容易分辨,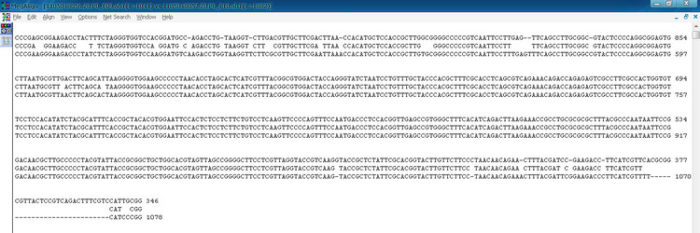
在序列上点右键,选择Alinment coloor设置显示参数,一般全选即可,这样可得到很直观的结果,最上面一栏显示相似性的一些信息。