

1 综述
PAML(将最大似然法用于系统发育分析)是一个用最大似然法来对DNA和蛋白质序列进行系统发育分析的软件包。
PAML 文档
除了这个手册,请注意以下资源:
PAML站点:http://abacus.gene.ucl.ac.uk/software/PAML.html 含有下载和编译程序的信息。
PAML FAQ页面:http://abacus.gene.ucl.ac.uk/software/pamlFAQs.pdf
PAML讨论组 http://www.rannala.org/phpBB2/,在这里你可以提交bug报告和提问。
PAML程序能做些什么
PAML软件包目前包含如下程序:baseml,basemlg,codeml,evolver,pamp,yn00,mcmctree,和chi2。Yang(2007)提供了一个简短的关于PAML中常用的模型和方法的综述。有本书(Yang 2006)描述了统计和计算的详细内容。分析的例子可以从软件包中找到。
l 比较和测试系统发育树(baseml 和codeml);
l 复杂替代模型中的参数估算,包括在位点和模型中用于对多个基因或位点划分进行结合分析的变化率模型。(baseml 和 codeml)
l 通过对实施的模型的对照进行似然率假设性检验(baseml,codeml,chi2);
l 基于全局或局部分子钟模型估算分歧时间(baseml 和 codeml);
l 使用核酸,氨基酸和密码子模型对祖先序列似然性(经验贝叶斯)重构(baseml 和 codeml);
l 用Monte Carlo模拟生成核酸,密码子和氨基酸序列数据集(evolver);
l 估算同义和非同义替代率和发现蛋白编码DNA序列中的正选择;
l 贝叶斯估算物种的分歧时间来合并化石尺度上的不确定性。
PAML的长处是它的一系列复杂替代模型。Baseml和codeml中使用的树搜索算法还都很简单,因此除非是非常小的数据如小于10个物种,否则你最好使用其他的软件包,如phylip,paup,或mrbayes来推断树的拓扑结构。你可以使用其他程序获得一系列的树并那它们作为使用树来通过baseml和codeml来评估它们。
Baseml和codeml:Baseml这个程序是使用最大似然法分析核酸序列。Codeml这个程序由两个旧程序合并构成,它们是codonml(Goldman和Yang(1994)的对蛋白编码DNA序列执行密码子替代模型)和aaml(用来执行氨基酸序列模型)。它们俩现在靠控制文件codeml.ctl中的变量seqtype来区分,1对应密码子序列,2对应氨基酸序列。这个文档中,我们使用codonml和aaml意味着codeml中的seqtype分别等于1和2。程序baseml,codeml和aaml使用相似的算法(最大似然法)来配合模型。三个程序主要的不同是马尔科夫模型中的进化单元不同,序列中被称为“位点”的分别是一个核酸,一个密码子或者一个氨基酸。马尔科夫模型用替代率(计算位点间不变或变化)来描述核酸,密码子或氨基酸间的替代。
Evolver:这个程序基于核酸,密码子和氨基酸替代模型模拟序列。它还具备一些其它的选项如生成随机树,计算树之间的分隔距离(Robinson 和 Foulds 1981)。
Basemlg:这个程序执行(连续的)Yang(1993)的gamma模型。当数据超过6或7个物种时它得到速度非常慢并且难执行。作为替代应使用Baseml中的离散gamma模型。
Mcmctree:这个执行的是Yang和Rannala(2006)和Rannala和Yang(2007)的bayesian MCMC算法来估算物种的分歧时间。
Pamp:这个执行的是Yang和Kumar(1996)的基于简约的分析。
Yn00:这个执行的是Yang和Nielsen(2000)用来成对估算蛋白编码DNA序列中同义和非同义替代率(ds 和 dn)的方法。
Chi2:这个用来执行似然率测试。它计算chi平方临界值。你可以将它同真实数据的检验统计相比较来确定检验在5%和1%的水平上是否显著。键入程序名chi2来运行这个程序。当你输入检验统计值和d.f时这个程序也能计算P值。通过键入chi2 p运行。
PAML不能做什么
有些事情你可能期望一个系统发育软件包应该做,但PAML不能做。这儿有部分列表,希望能帮你避免浪费时间。
序列对齐:你应该使用其它的一些程序,如Clustal或者TreeAlign来自动对齐序列或者手动对齐,或者从其它一些如BioEdit和GeneDoc这样的程序中得到帮助。手动调整似乎没有达到可以完全信赖的成熟的阶段,那么如果有可能,你应当总是手动调节。如果你在基因组范围内构建成千上万的对齐,你应该实行一些质量控制,计算一些序列分歧的评估来作为对齐的不可信度的指标。对于编码序列,你应当对齐蛋白序列并且基于蛋白的对齐来构建DNA的对齐。值得注意的是在baseml和codeml(如果cleandata=1)中,对齐的空格是作为缺失的数据来对待的。如果cleandata=1,所有的模糊位点和空格都会被移除。
基因预测:codeml中基于密码子的分析(codeml seqtype=1)假定序列是从外显子开始对齐,序列的长度是3的倍数,序列中的第一个核酸是编码的第一个位置。内含子、间隔和其它非编码区域必须被移除并且编码序列必须在运行程序前对齐好。这个程序不能直接运行从GenBank下载回来的序列,即使这里含有CDS的信息。它也不能预测出编码区域。
在大规模数据集中树搜索:如先前提到的,你应该使用其它的程序来获得一棵树或一些候选树,并使用这些树作为使用树来配合其它软件包中不可用的模型。
2编译和多系统下运行PAML程序
PAML使用旧式的命令行界面。你可以从PAML网站下载存档,典型的以PAML*.*tar.gz命名,在你硬盘上解压这些文件。这是一个针对所有平台的文件。可执行文件是针对windows,对于UNIX和MAC OS X,你需要在运行前先编译他们。
Windows
软件包中包含windows的可执行文件。
1.
到PAML的站点http://abacus.gene.ucl.ac.uk/software/paml.html下载最后的存档并保存在你的硬盘上。解压,如,使用WinZip,存档在一个文件夹里,如D:\software\paml\.记住文件夹名称。
2.
启动一个“命令行”,从“开始 – 程序 – 附件”。也可以“开始 – 运行”并键入cmd,点击ok。你可以右击标题栏来改变窗口的字体,颜色,大小等。
3.
改变路径到paml的文件夹。如下:
D:
d: \software\paml
dir
4.
注意,windows的命令和文件名是大小写不敏感的。文件夹 src 包含源文件。example各种例子文件。bin 文件夹包含windows的可执行文件。你可以使用windows浏览器来查看这些文件。在当前文件夹下,使用默认的控制文件baseml.ctl运行baseml程序。命令行大概如下。
D:\software\paml4\bin\baseml
Baseml将读取当前目录下的默认控制文件baseml.ctl并根据它的参数来执行分析。现在我们可以输出一个baseml.ctl的副本,并用一个文本编辑器来查看相关的序列和树文件。类似地,你也可以运行codeml并查看它的控制文件codeml.ctl.
接下来你可以准备自己序列文件和树文件。控制文件和其它输入文件都是纯文本文件。一个常见的错误是由于UNIX和windows对回车和换行的不同处理方式造成的。如果你使用MS word来准备这些输入文件,你应该保存为“以换行结尾的文本”或“不带换行的文本”。有时只有两者中的一种有用。不要将文件保存成word文档。我在附录B的“克服windows的困扰”一节中收集了一些注意事项。如果你坚持双击,你可以打开windows浏览器,复制可执行文件到你包含控制文件的文件夹,然后双击运行。
UNIX
软件包里没有提供UNIX的可执行文件,因此你必须使用软件包src文件夹中的源文件自己编译它们。值得注意的是UNIX命令行和文件或文件夹名称是大小写敏感的。下面是假设你在UNIX提示符下。
1.
到PAML站点http://abacus.gene.ucl.ac.uk/software/paml.html下载最后的文件并保存在硬盘上。使用gzip解压。命令如下:
gzip –d paml4.tar.gz xf paml4.tar
2.
你可以使用 ls 查看文件夹中的文件。删除bin文件夹下的windows可执行文件(.exe文件)。然后切换到src文件夹下用make编译。
ls -lF bin (列出bin文件夹下的.exe文件)
rm –r bin/*.exe
cd src
make
ls -lF
rm *.o
mv baseml basemlg codeml pamp evolver yn00 chi2 ../bin
cd .. bin/codeml
4.
这些命令编译程序并生成可执行文件baseml,basemlg,codeml,pamp,evolver,yn00,和 chi2。你可以用ls命令查看到。然后删除中间的目标文件*.o,然后移动编译好的可执行文件到PAML的主文件夹bin文件夹。用cd命令切换到PAML主文件夹并使用默认的控制文件codeml.ctl运行codeml.你可以输出codeml.ctl的副本并查看它(主要的结果文件mlc)。
如果编译(make命令)文件不成功,你可能必须在使用make命令前打开并编辑makefile文件。例如,你可以将cc改变为gcc 和-fast到-O3或-O4。如果这些都不起作用,查看scr文件夹中的readme.txt文件中的编译指令。你可以复制编译命令到命令行。例如:
cc –o baseml baseml.c tools.c –lm
cc –o codeml codeml.c tools.c -lm
将使用C编译器cc编译 baseml 和 codeml。然而这种情况下代码的优化没有被开启。你需要使用编译器切换到代码优化,如,
cc –o codeml –O3 codeml.c tools.c -lm
最后,如果你当前的文件夹不在你的搜索路径,你必须添加./到可执行文件的名字前面甚至可执行文件在你的当前工作目录时也是如此。即用./codeml 代替 codeml 来执行codeml。
Mac OS X
Mac OS X 如同 UNIX,你需要按照上面UNIX的说明来做。打开一个命令终端(应用 – 工具 – 终端)并且从终端编译和运行程序。你 cd 到 paml/src 文件夹并查看 readme.txt 或 makefile 文件。参照上面。如果你键入命令 gcc 或 make 并得到“命令未找到”的错误,你必须从Apple的站点http://developer.apple.com/tools/下载Apple Developer’s Toolkit。在FAQ里有一些MAC OS X或 UNIX下运行程序所要注意的内容。
我已停止发布MAC OS 9以前的老版本执行程序。
运行程序
如同上面指出的,你可以通过命令行键入它的名字来运行一个程序。你需要知道你的序列文件,树文件和控制文件在哪个文件夹,这与你的工作文件夹有关。如果不熟练,你可以拷贝这些可执行文件到包含你数据的文件夹。根据模型的使用,codeml可能需要一些数据文件诸如 grantham.dat,dayhoff.dat,jones.dat,wag.dat,mtREV24.dat,或 mtmam.dat。因此你可能也要复制这些文件。
这个程序产生一些名字如rub,lnf,rst,或 rates的结果文件。不要将这些名字用于自己的文件,否则他们将被覆盖。
示例数据
Examples文件夹包含许多例子数据。它们被作为原始文件来测试新的方法,我将他们包含进来以便你可以重复出我们文档中的结果。对齐的序列,控制文件和详细的说明文件也包含在内。它们可以帮助你获得熟悉的输入数据格式和带有解释的结果,也能帮助你发现程序中的bug。如果你对个例的分析感兴趣,那么拷贝一个带有描述方法和分析例子的文件来重复已公布的结果。这非常重要,因为这个手册只写出和描述了程序控制文件中变量的意义,但没有清楚的解释如何针对个例分析设定控制文件。
examples/HIVNSsites/: 这个文件夹包含Yang等人(2000b)的HIV-1 env V3 区域分析的例子文件。这个数据用来演示文档中描述的NSsites模型,也就是,氨基酸位点中变量ω比值的模型。这些模型被Yang和Swanson(2002)称为 “random-sites”模型。这是由于我们在推理中不知道哪些位点是高度保守并承受正选择的。它们也称为“fishing-expedition”模型。这些数据集是Yang等人(2000b)中分析的第十组数据,结果在该文章的表格12中。参看文件夹中的说明文件。
examples/lysin/:这个文件夹包含的是Yang, Swanson & Vacquier (2000a) 和 Yang and Swanson (2002)对25种鲍鱼的精子细胞溶解酶基因的分析数据。这个数据集是为了演示“random-sites” (Yang, Swanson & Vacquier 2000a)模型和“fixed-sites”(Yang and Swanson 2002)模型的。
后面的这篇文章中,我们使用了结构信息来区分细胞溶解酶中“埋入”的和“暴露”的氨基酸位点信息。并指定和估算两个不同分区的ω比值。假设就是暴露在表面的位点有可能承受正选择。参见文件夹中的说明文档。
examples/lysozyme/:这个文件夹包含的是Messier 和 Stewart(1997)及Yang(1998)重分析的灵长类溶菌酶c基因的数据。这是为演示codon模型,它能够对树的不同枝指定不同的ω比值,对沿着世系进行的正选择测试非常有效。这个模型有时也叫做branch模型或branch-specific模型。Yang(1998)的大小数据集都包含在内。这些模型需要用户标出树种的枝,说明文件和所包含的树文件里对格式进行了非常详细的解释。也可以参看“树文件和树拓扑结构的表示方法”一节中关于指定枝和节点的部分。
Yang 和Nielsen(2002)也使用了溶菌酶数据来执行被称为“branch-sites”的模型,它允许ω比值在世系和位点中都可以变化。参见学习如何运行这个模型的说明文件。
examples/MouseLemurs/: 这个文件夹包含的是Yang和Yoder(2003)通过 mtDNA比对来分析和估算老鼠与狐猴的分歧时间的数据。这个数据集用来演示如何通过最大似然法按照全局和局部分子钟估算分歧时间。文档中描述了更复杂的同时使用多个校准节点的模型的使用方法,对多个基因(或位点分区)进行分析来解释各组分枝间的不同和变化的比值。说明文件中解释了输入数据的格式和模型的详细设定方案。说明文件2中解释了Yang(2004)中对特别比值的平滑过程。
examples/mtCDNA/: 这个文件夹包含的是线粒体基因组中12个编码蛋白基因的同侧序列的比对结果。这些是Yang, Nielsen和Hasegawa(1998)用于对7种猿的密码子和氨基酸替代模型的分析数据。这个是文章中提到的小数据集,用来配合氨基酸替代率的mechanistic和empirical模型,也可以用于密码子替代的mechanistic模型。这个模型可用于测试保守的和进化的氨基酸替代率是否相等。详细的参看说明文件。
examples/TipDate/:这个文件夹包含的是Rambaut(2000)在TipDate模型中使用的例子数据。用于时间已知的病毒序列。说明文件解释了如何使用baseml来配合TipDate模型,用在一个不同时间的序列的全局的时钟上。局部时钟模型也可以应用。如何做请参看examples/MouseLemurs/文件夹。注意,我们使用符号@在序列名称前作为序列测定数据的前缀。文件可以使用Ranmbaut的TipDate程序阅读,但是我们软件包中的文件在送入baseml前需要进行一些编辑。
软件包中也包含了一些其它的数据,下面是详细内容。
brown.nuc and brown.trees: Brown 等人(1982)的895-bp的mtDNA数据。它们被Yang(1994)等人和Yang(1994b)用来测试位点中变化比值的模型。
mtprim9.nuc and 9s.trees:由来自9个灵长类的888个对齐的位点构成的线粒体片段。被Yang(1994a)用于测试离散gamma模型和Yang1995测试自动离散gamma模型。
abglobin.nuc and abglobin.trees:连锁的α和β球蛋白基因。被Goldman和Yang(1994)用在对密码子模型的描述中。abglobin.aa是翻译的氨基酸序列的比对数据。
stewart.aa and stewart.trees:6种哺乳动物的溶解酶蛋白序列(Stewart等人,1987)。被Yang等人(1995a)用来测试重建原始氨基酸序列的方法。
3数据文件格式
序列数据文件格式
参看软件包中示例数据文件(.nuc .aa和.nex)。只要你获得其中一种格式的数据,PAML程序就可以读入它。默认的格式是(Felsenstein 2005)PHYLIP软件包中的PHYLIP格式。PAML有限的支持PAUP和MacClade中使用的NEXUS文件格式。只有序列数据或树可以被读入,注释块被忽略。PAML不处理序列数据块中的注释块,因此请清除它们。
连续的和交替的格式
下面是一个PHYLIP(Felsenstein 2005)格式的例子。第一行包含物种的数目和序列长度(可能跟随着选项符)。对于密码子序列(codeml seqtype = 1),序列文件中的序列长度是指核酸的数目而不是密码子的数目。序列文件中的选项只能是I,S,P,C和G。序列既可能是交替格式(选项I,示例数据文件abglobin.nuc),或者连续的格式(选项S,示例数据文件brown.nuc)。默认选项是S,因此你并不需要指定它。选项G用于多基因数据的结合分析,见下面解释。这面时一个连续型示例数据集。它有4条60个核酸(或20个密码子)的序列。
4 60
sequence 1
AAGCTTCACCGGCGCAGTCATTCTCATAAT
CGCCCACGGACTTACATCCTCATTACTATT
sequence 2
AAGCTTCACCGGCGCAATTATCCTCATAAT
CGCCCACGGACTTACATCCTCATTATTATT
sequence 3
AAGCTTCACCGGCGCAGTTGTTCTTATAAT
TGCCCACGGACTTACATCATCATTATTATT
sequence 4
AAGCTTCACCGGCGCAACCACCCTCATGAT
TGCCCATGGACTCACATCCTCCCTACTGTT
物种/序列名称:物种和序列名字中不要使用下面的特殊符号¨, : # ( ) $ =〃,它们用于特殊的目的可能会引起程序的混乱。@符号可以作为序列名称的一部分或结尾来指定序列测试的时间,例如virus1@1984。@符号被认为是名称的一部分表示这个序列是在1984年测试的。物种名称的最大字符数在主程序baseml.c和codeml.c的开头指定的。在PHYLIP中,物种名是10个字符,我发现经常受限制,因此我将默认值设为30。为解决这个问题,PAML中认定两个连续的空格作为物种名的结束符,以避免物种名要30个(或10个)字符。为遵守这一规则,你不能再一个物种名中使用两个连续的空格。例如上面的数据也可以用下面的格式。
4 60
sequence 1 AAGCTTCACCGGCGCAGTCATTCTCATAAT
CGCCCACGGACTTACATCCTCATTACTATT
sequence 2 AAGCTTCACCGGCGCAATTATCCTCATAAT
CGCCCACGGACTTACATCCTCATTATTATT
sequence 3 AAGCTTCACC GGCGCAGTTG TTCTTATAAT
TGCCCACGGACTTACATCATCATTATTATT
sequence 4 AAGCTTCACCGGCGCAACCACCCTCATGAT
TGCCCATGGACTCACATCCTCCCTACTGTT
如果你想使文件同时被PHYLIP和PAML识别,你需要限定名称的字符数为10并且至少用两个空格来分隔名字和序列。
在一个序列中,T,C,A,G,U,t,c,a,g,u都被识别为核酸序列(在baseml,basemlg和codonml中),标准的单字符编码(A, R, N, D, C, Q, E, G, H, I, L, K, M, F, P, S, T, W, Y, V及小写形式)被识别为氨基。模糊字符(未定的核酸或氨基酸)也允许使用。三个特殊字符“.”“-”“?”做如下解释:圆点表示在第一个序列中相同的字符,破折号表示一个比对的空位,问号表示未确定的核酸或氨基酸。序列中的非字母符号,诸如><!ˇ”$%&^[](){}0123456789被忽略并且作为标志自由使用。行不需要等长并且你可以在一行中输入整个序列。
Baseml和codeml中处理模糊字符和比对空位的方法依赖控制文件中的cleandata变量。
在最大似然分析中,当cleandata = 1时,如果一条序列的位点上包含了一个模糊的字符,那么分析前,该位点将被从所有的序列中移除。当cleandata = 0时,模糊字符与比对空位都将被作为模糊字符对待。在成对的距离计算中(输出中的下对角距离矩阵),当cleandata = 1时,表示“完全删除”,所有序列中的全部模糊字符和比对空位都被移除,当cleandata = 0时,表示“成对删除”,只有都为缺失字符时才被成对的移除。
PAML程序中没有针对插入和缺失的模型。因此,一个比对空位会被作为模糊对待(也就是一个问号?)。注意,在密码子序列中移除任何核酸意味着移除整个密码子。
注释可能位于序列文件的末尾并且被程序忽略。
选项G:这个选项是针对不同数据集如多基因数据或三个密码子位置的数据的结合分析。序列必须连续并且选项用来指定每个位点来自哪些基因或密码子。
这个选项有三种格式。第一种格式用下面一个序列文件的摘录来说明。示例数据时Brown等人(1982)年的数据,来自线粒体基因组的895bp的一个片段,编码两个蛋白(ND4和ND5)的末尾部分和中间的三个tRNAs。序列中的位点分成4类:3个密码子位置和tRNA编码区域。文件第一行包含选项符G。下面的行里包含位点标记,序列中的每个位点一个(在codonml中试每个密码子一个)。位点标记指出每个位点是哪一类。如果有g类,标记就是1,2,……,g,并且如果g > 9,标记需要通过空格来分隔。标记的总数目必须等于每个序列中的位点的总数。
5 895 G
G 4
3
123123123123123123123123123123123123123123123123123123123123
123123123123123123123123123123123123123123123123123123123123
123123123123123123123123123123123123123123123123123123123123
123123123123123123123123123123123123123123123123123123123123
123123123123123123123123123123123123123123123123123123123123
123123123123123123123123123123123123123123123123123123123123
123123123123123123123123123123123123123123123123123123123123
1231231231231231231231231231231231231
444444444444444444444444444444444444444444444444444444444444
444444444444444444444444444444444444444444444444444444444444
444444444444444444444444444444444444444444444444444444444444
444444444444444444
123123123123123123123123123123123123123123123123123123123123
123123123123123123123123123123123123123123123123123123123123
123123123123123123123123123123123123123123123123123123123123
12312312312312312312312312312312312312312312312312312312312
Human
AAGCTTCACCGGCGCAGTCATTCTCATAATCGCCCACGGACTTACATCCTCATTACTATT
CTGCCTAGCAAACTCAAACTACGAACGCACTCACAGTCGCATCATAATC……..
Chimpanzee
………
第二种格式用于连续的多基因数据,如下面例子数据中所示。这个序列由来自4个基因的1000个核酸构成,基因1,2,3,4分别提供了100,200,300和400个核酸。基因的“长度”必须用G开头的行中指定,如例子中的第二行。(这个要求准许程序确定两种格式中的哪种被使用。)所有基因的长度和必须等于组合成的序列中的核酸,氨基酸或密码子的数目,以便用于baseml(或basemlg),aaml,和codonml。
5 1000 G
G 4 100 200 300 400
Sequence 1
TCGATAGATAGGTTTTAGGGGGGGGGGTAAAAAAAAA…….
第三种格式仅用于蛋白编码DNA序列(针对baseml)。你在第一行用选项符GC取代G。程序会在基于核酸的分析中有区别的对待三个密码子位置。这里假定序列的长度正好是3的倍数。
5 855 GC
human GTG CTG TCT CCT …
密码子序列中的选项G(codeml seqtype = 1)。这个格式类似于baseml中的选项,但是值得注意的是序列的长度是核酸的数目,而基因的长度是密码子的数目。这里容易引起混淆。下面是一个例子:
5 300 G
G2 40 60
这个数据里包含5个序列,每条含300个核酸(100个密码子),它分成两个基因,第一个基因含40个密码子,第二个基因含60个密码子。
位点模式计算
序列比对结果也可以输入到位点模式的格式和含有这些位点模式的位点计算中。这个格式用输入文件第一行中的选项“P”来指定。用下面例子来说明。这里有三个序列,8个位点模式,用“P”指明数据是位点模式而不是位点。“P”选项与交替格式的选项“I”和连续格式的选项“S”用法相同。比对结果下面的8个数字是上面具有8个模式的位点的数目。例如,有100位点,所有的3个物种都是G,有200个位点所有三个物种都是T,等等。总共有100+200+40+……+14 = 440个位点。
3 8 P
human GTACTGCC
rabbit GTACTACT
rat GTACAGAC
100 200 40 50 11 12 13 14
这个例子用于baseml和basemlg程序,是针对基于核酸的分析。指定多个基因(位点模式),也可以将选项G和P合起来用。
3 10 PG
G 2 4 6
human GTTA CATGTC
rabbit GTCA CATATT
rat GTTA CAAGTC
100 200 40 50 120 61 12 13 54 12
这里有一个位点模式和2个基因(位点模式)。前4个模式是第一个基因,接下来的6个模式是第二个基因,共计10个位点模式。分区1有40个位点是AAA(三个物种中都是核酸A),分区2中有61个这样的位点。
蛋白序列也可以使用同样的格式(codeml seqtype = 2),就是将上例中的核酸替换成氨基酸。
对于密码子序列(codeml seqtype = 1),格式如下。有3个物种和9个位点模式6个位点是第一位点模式(三个物种中都是密码子GTG)。值得注意的是27 = 9 * 3。程序需要你用密码子位点模式数量的3倍。这有点奇怪,但是符合连续的或交替的序列格式,这里的序列长度指的是核酸数目而不是密码子的数目。(最初我这么做是为了可以让同一个文件能被基于核酸分析的baseml和基于密码子分析的codeml所识别。)
3 27 P G
human GTG CTG TCT CCT GCC GAC AAG ACC
rabbit … … … G.C … … … T..
rat … … … ..C ..T … … …
6 1 1 1 1 4 3 1 1
指定多个基因的密码子位点模式,参见下面的例子。
3 27 P G
G 2 4 5
human GTG CTG TCT CCT GCC GAC AAG ACC
rabbit … … … G.C … … … T..
rat … … … ..C ..T … … …
6 1 1 1 1 4 3 1 1
总计9个密码子位点模式,基因1含有前4个模式,基因2含有后5个模式。
此外,选项变量P能同选项变量I或S结合使用。PI意味位点模式使用的是交替的格式,而PS代表位点模式使用连续的格式。P不带有I或S则默认使用的是连续格式。一行里放入整个序列的全部位点模式同时符合I和S格式,因此不需要指定I或S。
如果你运行baseml和codeml来读取连续或交替的序列格式,输出将包含一个分隔格式的输出。参见“输出位点模式计算”。如果选择位点模式需要花很长时间,你可以将这部分移动到一个新文件中,稍后作为替代的读入文件。注意下面使用P格式的限制。
这个选项有一些限制条件。这个选项禁止了一些输出,包括原始序列的重构和对位点后估算的比值,它可以通过RateAncestor = 1获得。其次,一些计算需要序列长度,我设为位点模式频次的总和。如果位点模式频次不能通过位点计算但是有被位点模式代替的可能,计算中包括序列长度将造成错误。
例如,计算包括针对MLEs的SEs,在codonml中位点S和N的数目。可能使用这个选项。有时我用evolver模拟非常长的序列(>1M个位点),当我用最大似然迭代只用了几秒以及我想将同样的数据用来运行多个模型时,而它可能要用几分钟甚至几小时的时间将位点转化成模式,这是非常让人恼火的。类似的情况还有对大规模基因组数据中超过100Mb位点的长序列进行分析的时候。这种情况下你可以运行一次baseml或codeml,然后从输出文件中复制模式计算结果到一个数据文件。下一次,你运行程序时可以读入新的文件。这个方法可以使程序跳过计算位点模式的步骤。另一种情况是基于模型计算位点模式的概率并且在分析时使用一个错误的树来读入概率来看正确的树是否被重建。用这个方法你可以检查树的重构方法是不是一致的。参见Debry(1992)和Yang(1994c)的分析。(我需要用密码子来输出位点模式概率。)
树文件格式和树拓扑结构的表示方法
当runmode = 0 或 1时需要使用树结构文件。文件名通过适当的控制文件指定。典型的树拓扑结构是用圆括号指定,虽然有时也可能用下面叙述的分枝方式表示。
圆括号:第一种是用熟悉的圆括号来表示,在大多数系统发育软件中通用。物种可以用他们的名称或者当前序列数据文件中一致的序号来表示。如果使用了物种名,你必须与他们序列文件中的名称严格匹配(包括空格和特殊符号)。允许使用枝的长度。下面是一个含有四个物种的树结构文件(文件中的顺序为human,chimpanzee,gorilla和orangutan)。第一个树为星型树,接下来的四个树也是一样。
4 5 // 4 个物种, 5 棵树
(1,2,3,4); // 星型树
((1,2),3,4); // 物种1和2聚在一起
((1,2),3,4); // 超过9个物种时需要使用逗号
((human,chimpanzee),gorilla,orangutan);
((human:.1,chimpanzee:.2):.05,gorilla:.3,orangutan:.5);
如果树有枝长度,baseml和codeml允许使用枝长度作为最大似然迭代的起始值。
你使用有跟树或无根树取决于你的模型,例如,估算一个分子钟。没有钟(clock = 0),需要用无根树,如((1,2),3,4) 或(1,2,(3,4))。有钟或局域钟模型,需要有根树并且这两个树是不同的而且都不同于(((1,2),3),4)。在PAML中,有跟树在根部有分枝,而无根树在根部有三个或多个分支。树文件用PAUP和MacClade生成。PAML程序仅与PAUP或MacClade生成的树有限的兼容。首先,用来指定无根树的符号“[&U]”被忽略。你必须手动修改树文件使它在根部含有3个分支以便PAML将它识别为无根树。例如将(((1,2),3),4)改为((1,2),3,4)。其次,关键词“Translate”被PAML忽略,它假定树文件中的序列顺序与序列数据文件中的序列顺序严格一致。
枝或节点标签:在运行baseml和codeml的一些模型时允许将树中的一些枝分组,并且对它们分配感兴趣的参数。例如:baseml和codeml中的局部分子中模型(clock = 2或3),你可以分3个比值组,分别是低,中,高。并且branch-specific codon模型(codonml model = 2 或 3)允许不同的分枝组拥有不同的ω值,因此被称为“two-ratios”和“three-ratios”模型。所有这些模型需要在树的枝或节点上做标签。除使用“#”而不是“:”以外,枝标签与枝长的指定方式一样。枝标签是从0开始的连续整数,这是默认的并且不需要指定。例如下面的这个树
((Hsa_Human, Hla_gibbon) #1, ((Cgu/Can_colobus, Pne_langur), Mmu_rhesus), (Ssc_squirrelM, Cja_marmoset));
来自文件 examples/lysozyme/lysozyme.trees。它带有一个枝标签来匹配模型中枝的不同ω比值。对human和gibbon的内部祖先枝含有比值ω1,而其它枝(默认标签为#0)拥有背景比值ω0。这个是针对溶菌酶基因的小数据集来配合表1C中的模型(Yang 1998)。参见examples/lysozyme/文件夹中的说明文档。
对于一个大的树,你可能想标记一个进化枝内全部分枝。出于这个目的,你可以使用进化枝标签$。$是替代Δ,Δ更适合做进化枝的符号,但很多键盘上没有它。因此(clade) $2等同于用#2标记进化枝内所有的节点或枝。下面的两个树也同样。
(((rabbit, rat) $1, human), goat_cow, marsupial);
(((rabbit #1, rat #1) #1, human), goat_cow, marsupial);
这里有一些关于嵌套进化枝标签的规律。符号#的优先级高于$,树顶端的进化枝标签优先于接近根处的祖先节点的进化枝标签。下面的两棵树也是一样。第一个树中$1适用于整个胎盘哺乳动物的进化枝(除人类世系外),$2适用于兔与鼠的进化枝
((((rabbit, rat) $2, human #3), goat_cow) $1, marsupial);
((((rabbit #2, rat #2) #2, human #3) #1, goat_cow #1) #1, marsupial);
注意,使用这个规律,你是否使用$0可能会有些不同。例如
((a, b) $0, (c, d)) $1;
#0标记了左侧的三个枝(a,b,以及a和b)而其它枝为#1。虽然#1是针对树中的所有分支的。
((a, b), (c, d)) $1;
我发现使用Rod page (1996)的TreeView软件可以很方便的创建树文件并且检查树和标签是否正确。上面作为例子的树都可以被TreeView识别。在TreeView X中,你必须使用单引号来标记。如下:
((((rabbit ‘#2′, rat ‘#2′) ‘#2′, human ‘#3′) ‘#1′, goat_cow ‘#1′) ‘#1′, marsupial);
这样,这个树就可以同时被TreeView和TreeView X识别(baseml和codeml也一样)。
注意,TreeView和TreeView X不接受顶端或顶端分支的标签,可能会将标签解释为序列名称的一部分。另一个可以创建和浏览枝与节点标签的程序是Andrew Rambaut的TreeEdit,可用用在MAC上。我没怎么用过它。
分歧时间符号@:化石刻度的信息通过使用符号@来指定。在baseml和codeml的钟与局域钟模型中有使用。参看examples/MouseLemurs/ 文件夹的说明文档。下面的例子中human-chimpanzee的分歧时间被设定为5MY。
((gorilla, (human, chimpanzee) ‘@0.05′), orangutan);
贝叶斯时间程序mcmctree校准信息参见后面的描述。
树拓扑结构的枝表示方法:PAML中使用的树的拓扑结构的第二种表达方法是列举它的枝,每一个枝用它的起始和终止节点来表示。这个表达方式也用于估算枝长度的结果输出文件中,你也可以将它用作树文件。例如,树((1,2),3,4)可以用列举它的5个枝来指定:
5
5 6 6 1 6 2 5 3 5 4
树中的节点用连续的自然1,2,…s数来表示数据中已知的s个序列,顺序与数据中一致。一个标签大于数值s的内部节点,它的序列是未知的。因此,在上面的树中,节点5是节点6,3和4的祖先节点,节点6是节点1和2的祖先节点。
这种表示法便于指定数据中的一些序列直接是其它序列的祖先的树。例如,下面是5个序列4个分支的一棵树,序列5是序列1,2,3,4的共同祖先。
4
5 1 5 2 5 3 5 4
警告:我没有对baseml和codeml中的全部模型使用这种树的表达方法。如果你使用这种表达方式,需要仔细的测试你的程序。如果他没有工作,你可以告诉我一边我来订正它。
4 baseml
核酸替代模型
对核算替代模型中马尔科夫模型的详细描述,参见whelan(2001),Felsenstein(2004)或Yang(2006 第1章)。
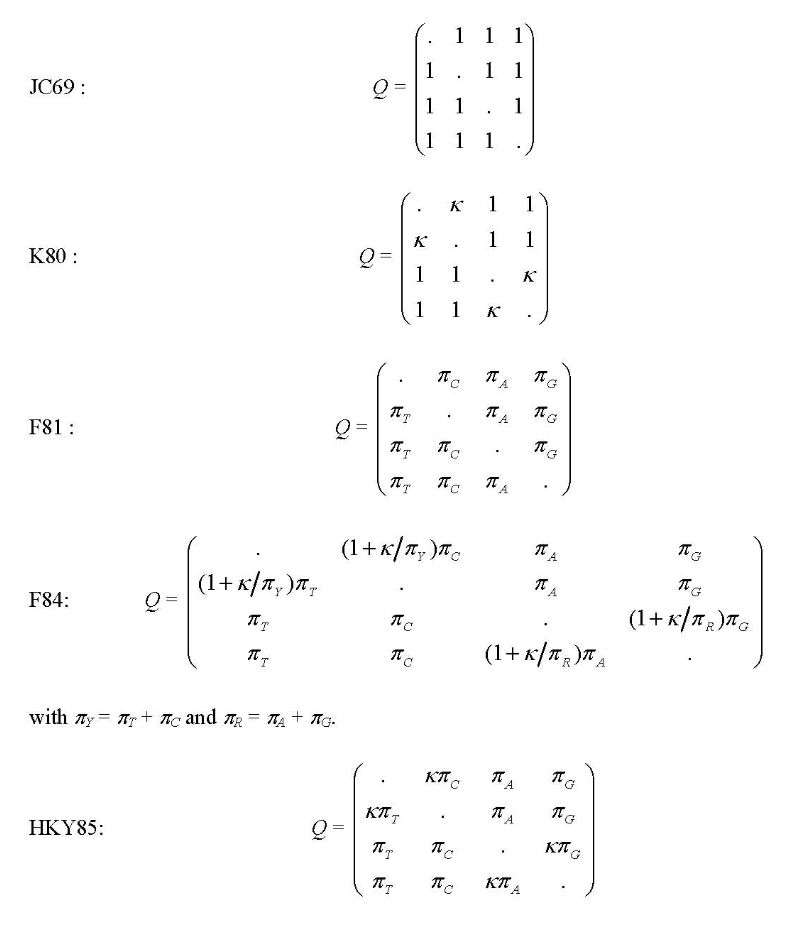
PAML中使用的模型包括JC69(Jukes 和 Cantor 1969),K80(Kimura 1980),F81(Felsenstein 1981),F84(Felsenstein,DNAML 1984,PHYLIP 2.6版),HKY85(Hasegawa等 1984;Hasegawa 等 1985),Tamura(1992),Tamura 和 Nei(1993),和REV也称为GTR用于general-time-reversible(Yang 1994b;Zharkikh 1994),比值矩阵用下面的参数表示。

控制文件
baseml默认的控制文件是baseml.ctl,下面是个例子。注意,等号两侧的空格是必需的,空行或以“*”开始的行作为注释对待。不需要的选项可以从控制文件中删除。变量的顺序也并不重要。
seqfile = brown.nuc * sequence data file name
outfile = mlb * main result file
treefile = brown.trees * tree structure file name
noisy = 3 * 0,1,2,3: how much rubbish on the screen
verbose = 0 * 1: detailed output, 0: concise output
runmode = 0 * 0: user tree; 1: semi-automatic; 2: automatic
* 3: StepwiseAddition; (4,5):PerturbationNNI
model = 5 * 0:JC69, 1:K80, 2:F81, 3:F84, 4:HKY85
* 5:T92, 6:TN93, 7:REV, 8:UNREST, 9:REVu; 10:UNRESTu
Mgene = 0 * 0:rates, 1:separate; 2:diff pi, 3:diff kapa, 4:all diff
* ndata = 1 * number of data sets
clock = 0 * 0:no clock, 1:clock; 2:local clock; 3:CombinedAnalysis
fix_kappa = 0 * 0: estimate kappa; 1: fix kappa at value below
kappa = 2.5 * initial or fixed kappa
fix_alpha = 1 * 0: estimate alpha; 1: fix alpha at value below
alpha = 0. * initial or fixed alpha, 0:infinity (constant rate)
Malpha = 0 * 1: different alpha’s for genes, 0: one alpha
ncatG = 5 * # of categories in the dG, AdG, or nparK models of rates
fix_rho = 1 * 0: estimate rho; 1: fix rho at value below
rho = 0. * initial or fixed rho, 0:no correlation
nparK = 0 * rate-class models. 1:rK, 2:rK&fK, 3:rK&MK(1/K), 4:rK&MK
nhomo = 0 * 0 & 1: homogeneous, 2: kappa for branches, 3: N1, 4: N2
getSE = 0 * 0: don’t want them, 1: want S.E.s of estimates
RateAncestor = 0 * (0,1,2): rates (alpha>0) or ancestral states
Small_Diff = 1e-6
* cleandata = 1 * remove sites with ambiguity data (1:yes, 0:no)?
* icode = 0 * (RateAncestor=1 for coding genes, “GC” in data)
* fix_blength = 0 * 0: ignore, -1: random, 1: initial, 2: fixed
method = 0 * 0: simultaneous; 1: one branch at a time
控制变量的描述如下。
seqfile,outfile 和 treefile:分别指定序列数据文件的名称,主要结果文件和树结构文件。文件名中不能含有空格。文件名中尽量避免使用特殊字符,它们在系统中可能有特殊的含义。
noisy:控制屏幕上输出内容的多少。如果模型包含大量计算,你可以为noisy选择一个较大的数值以免寂寞。verbose控制结果文件中输出多少内容。
runmode = 0 表示树拓扑结构的评价在树结构文件中指定,runmodel = 1或2表示通过star-decomposition算法进行启发式树搜索。runmodel = 2,算法从星型树开始,如果runmodel = 1,程序将从树结构文件中读取一个多杈树并试图估算一个最好的分杈树与它相匹配。runmodel = 3表示逐步添加。runmodel = 4表示使用简约性算法获得起始树来进行NNI干扰,而runmodel = 5表示NNI干扰用的起始树是通过读取树结构文件得到的。树搜索选项做的不好,因此尽可能使用runmodel = 0。对于小数据集,逐步添加算法更有效些。
model:指定核酸替代模型。模型0,1,…,8分别表示模型JC69, K80, F81, F84, HKY85, T92, TN93, REV (也称为 GTR), 和 UNREST。注释参看Yang(1994 JME 39:105-111)。两个模型可以分别运行。model = 9是REV模型的特例,而model = 10是无限制模型的特例。格式如下面所示不需要过多解释。当model = 9或10时,同一行里包含的主要是指定模型的额外信息。中括号中的数字是自由参数比值的数字。例如,REV应该是5而UNREST应该是11。接在这些数字后面的是圆括号中的数字。输出文件中的比值参数将按照这里的顺序摆列。括号中没有提到的比值为1。当model = 9时,你只能指定TC或CT,但不能同时指定。当model = 10时,TC和CT是不同的。参见下面的例子和Yang(1994a)的注释。
model = 10 [0] /* JC69 */
model = 10 [1 (TC CT AG GA)] /* K80 */
model = 10 [11 (TA) (TG) (CT) (CA) (CG) (AT) (AC) (AG) (GT) (GC) (GA) ] /* unrest */
model = 10 [5 (AC CA) (AG GA) (AT TA) (CG GC) (CT TC)] /* SYM */
model = 9 [2 (TA TG CA CG) (AG)] /* TN93 */
Mgene:是结合序列文件中选项G用于多基因或多位点模式的结合分析(如三密码子位点)(Yang1996a;Yang 2006 116-8页)。这个模型也称为分隔模型。如果数据文件中没有G选项则选择0。
这里的描述只提到多基因的情况,但是它可以用于任何分割位点的策略,比如密码子位点的情况。类似的分割模型还有codeml中针对密码子或氨基酸序列的分析。
表1中总结了这些模型。最简单的模型假定基因中是完全均一的,不使用选项G,它可以将不同的基因连接成一个序列来处理(在控制文件中指定 Mgene = 0)。最普通的模型就是一个单独的分析,它对每个数据集(每个基因)采用同一个模型来完成任务,也可以通过指定Mgene = 1并结合带有G选项的数据文件来完成任务。覆盖不同基因的log似然值的总和在这里被认为是最常用模型的log 似然值。用于说明多基因的不均匀性方面的模型通过指定Mgene来与带有选项G的序列数据文件结合使用。Mgene = 0表示一个假设替代率不同但不同基因的核酸替代模式都相同的模型。Mgene = 2表示不同基因有不同的频率参数,但是速率参数相同(在K80, F84, HKY85 模型或TN93和REV模型中的速度参数)。Mgene = 3表示不同的速率参数和相同的频率参数。Mgene = 4表示不同的基因使用不同的速率参数和不同频率参数。参数和设定在表中有以HK85为例的总结。当假设位点和位点的替代速率是变化时,控制变量Malpha将指定是否所有的位点为一个gamma分布(Malpha = 0)或每个基因(或密码子模式)使用不同的gamma分布。
表1. 核酸替代率的分割模型的设定
| 序列文件 | 控制文件 | 基因中的参数 |
| 无 G | Mgene = 0 | 全部一致 |
| 选项 G | Mgene = 0 | k和π相同,cs不同(均衡的枝长) |
| 选项 G | Mgene = 2 | k相同,πs和cs不同 |
| 选项 G | Mgene = 3 | π相同,ks和cs不同 |
| 选项 G | Mgene = 4 | k,πs和cs都不同 |
| 选项 G | Mgene = 1 | 不同的k,πs,和不同(不均衡)的枝长 |
针对不同基因的不同cs表示对不同基因估算的枝长是均衡的。参数π代表均衡的核酸频率,它由观测到的频率估算而来(nhomo = 0)。HKY85中的转换/颠换比k可以分别被TN93或REV模型下的2或5个速率参数所替代。可以使用一种与方差分析非常相似的被称为差异分析的方法(McCullagh 和 Nelder 1989)得到的似然率来比较这些模型。
ndata:指定文件中分割数据的数量。这个变量用于模拟。你可以使用evolver来生成200份数据,然后设定ndata = 200并用baseml来分析。
clock指定模型涉及的种系中的速率是恒定的或变化的。
clock = 0表示从分枝到分枝没有时钟和速度是完全自由改变的。这个模型需要使用无根树。对于一个含有n个物种(序列)的分枝树,这个模型含有(2n-3)个参数(枝长)。clock = 1表示全局时钟,所有的枝具有相同的速度。这个模型有(n-1)个参数与分枝树中的(n-1)个内部节点相一致。因此,一个为比较两个模型的分子钟假设的测试需要d.f. = n-2。
clock = 2指定局部分子钟模型(Yoder和Yang 2000;Yang和Yoder 2003)。系统发育中的绝大多数枝与分子钟假设一致并有默认的速度(r0 = 1),但是一些预定义的分枝可能有不同的速度。分枝的速度通过在树文件中使用枝标签(#和$)指定。例如树(((1,2) #1, 3), 4)指定物种为1和2的祖先枝的速度为r1,而其它枝不需要指定,为默认的速度r0。用户需要指定哪些枝使用哪些速度,程序估算未知的速度(如上例中的r1;r0是默认速度)。当对分枝指定速度时你需要格外小心以确保模型可以估算全部速度;如果你指定太多的速度参数,尤其是对根周围的分枝,它可能无法估算全部的值而且你可能遇到是否可识别的问题。局域时钟模型的二叉树的参数数目是(n-1)加上分枝的额外速度参数的数目。上面那个4个物种的树中,你只有一个额外的速度参数r1,因此局域时钟模型含(n-1)+1 = n = 4个参数。对这棵树来说,非时钟模型有5个参数而全局时钟有3个参数。
clock = 3用于多基因或多分区数据的结合分析,它允许数据分区中,分枝的速度以不同的方式变化(Yang 和 Yoder 2003)。为解释数据分区中进化过程中的不同,你必须像控制文件(baseml.ctl 或 codeml.ctl)中的控制变量Mgene一样在序列文件中使用选项G。
阅读Yang和Yoder(2003)的工作和example/MouseLemurs/文件夹中的说明文档来重复论文中的分析过程。变量(= 5或6)也用来执行特殊速率的平滑过程(Yang 2004)。参见文件readme2.txt中的简介和文献中模型的详细说明。
当clock = 1,2,或3时,需要使用有根树。如果在树文件中使用符号@或=指定了化石尺度信息,计算的是绝对速度。通过这种方法可以指定多个校准位点,但只有点校准(这个点的年龄假设是已知无误的)可以接受而范围校准不能被接受。参见mcmctree程序的说明,它可以接受范围或其它分布的校准。如果序列含有时间,这个选项将适用于Andrew Rambaut的TipDate模型。
fix_kappa:指定K80,F84, 或HKY85中的k是否作为合适的值被给定或通过对数据的迭代来估算。如果fix_kappa = 1,另一个变量的值——kappa是已知值,而其它情况下kappa的值作为迭代的初始值来使用。变量fix_kappa和kappa对JC69或F81没有影响(它们不包括这些参数),或者TN93和REV(它们分别含有2和5个参数,全部从数据中估算)。
fix_alpha 和 alpha工作的方式大致相同,alpha针对跨越位点的变化的替代速率引用gamma分布的形状参数(Yang 1994a)。一个针对所有位点的单独速率的模型通过fix_alpha = 1和 alpha = 0(0代表无限)来指定,(离散的)gamma模型通过赋予alpha一个正值来指定,并且ncatG是离散gamma模型(baseml)中类别的数值。诸如5,4,8或10这样的值都是合理的。注意,离散gamma模型只有一个参数(α)时如同连续gamma模型,类型的数值不是一个参数。
不变位点模型在PAML程序中不能运行。我不喜欢这个模型在不变位点部分和gamma形状参数间生成一个强相关。它可以在PAUP和MrBayes中运行。
为推断独立位点的速率,适用RateAncestor = 1。见下面。如果你对计算这个速度有兴趣,使用一个大的类别数(如,ncatG = 40)或许有帮助。
模型的详细描述见Yang(1996b),Yang(2006)的第1章和Felsenstein(2004)。、
fix_rho 和 rho 工作方式类似并且涉及相邻位点上独立或相关的速率,rho是自动离散gamma模型(Yang 1995)的相关系数。针对位点的独立速率的模型以fix_rho = 1 和 rho = 0来指定;再选择alpha = 0表示针对所有位点的一个恒定速率。自动离散gamma模型通过对alpha和rho设定正值来指定。针对位点的恒定速率模型是α=∞ (alpha = 0)的(离散)gamma模型的特例,针对位点的独立速率模型是ρ= 0(rho = 0)的自动离散gamma模型的的一个特例。
nparK 指定针对变量和跨越位点的马尔科夫独立速率的非参数模式:nparK = 1 或 2 表示针对位点的独立速率的一些(ncatG)类别,nparK =3 或 4表示邻接位点上马尔科夫独立的速率;nparK =1 和 3有每个速率类别具有相等概率的限制,而nparK =2 和 4 没有这个限制(Yang 和 Roberts 1995)。变量nparK优先于alpha和rho。
nhomo仅针对baseml,并涉及同一替代模型中的频率参数。选项nhomo = 1适合一个均匀模型,但是通过最大似然迭代估算频率参数(πT,πC和πA;πG在频率总和为1中不是一个自由参数。)。这适用于F81,F84,HKY85,T92(这种情况下πGC是一个参数),TN93,或REV模型。通常(nhomo = 0)这些通过观察到的频率的平均值来估算。在两种情况下(nhomo = 0 和 1),你需要为碱基频率计算三个(或对T92计算一个)自由参数。
选项 nhomo = 3,4,和5仅用于F84,HKY85 或 T92。它们适合Yang ,Roberts(1995)和Galtier,Gouy(1998)的非均匀模型。核酸替代率通过变量model指定并且用于F84,HKY85或T92模型之一,考虑到不同序列中的碱基频率是不相等的,对于树中不同分枝的速度矩阵使用不同的频率参数。使用有根树时,根的位置对似然率有影响。由于参数丰富,模型仅用于小型树除非你有极长的序列。Yang和Roberts(1995)适用HKY85或F84模型并用三个独立的频率参数来描述替代模式,而Galtier和Gouy(1998)使用T92替代模型并只使用了GC含量πGC,碱基频率按πT=πA=(1-πGC)/2和πC = πG = πGC/2来得到。选项nhomo = 4为根分配一个频率参数集作为根的初始碱基频率,同样对树中的每个枝也分配一个频率参数集。如果使用的替代模型为F84或HKY85那么这就是Yang和Roberts(1995)工作中的N2模型,又或者使用T92替代模型的话,那么它就成为Galtier和Gouy(1998)工作中的模型。
选项nhomo = 3为每个顶端分枝使用一个碱基频率集,为树中所有内部分枝使用一个频率集,以及为根分配一个频率集。这就是Yang和Roberts(1995)工作中指定的N1模型。
选项nhomo = 5允许使用者指定需要使用多少个频率参数集和哪个节点(分枝)对应哪个集合。对根指定的集合作为根的初始碱基频率,而任何其它节点的参数集则作为从枝到节点的替代矩阵的参数。你在树文件(参见上面的“树文件格式和树拓扑结构的表示方法”部分)中用枝(节点)标签告诉程序每个枝应该使用那个集合。不需要指定默认的集合(0)。因此,例如nhomo = 5并且树文件中的物种1,2,3,4和5作为顶端分枝,6作为根,所有内部分枝(节点)都为默认的集合0。这相当于nhomo = 3。
((((1 #1, 2: #2), 3 #3), 4 #4), 5 #5) #6;
nhomo = 3,4,5的输出见“分枝的碱基频率参数和节点的频率”。对每个节点列出了两个频率的集合。第一个集合是参数(用于从枝到节点的替代速率矩阵),第二个集合是节点上预期的碱基频率,从模型计算而来(Yang 和Roberts 1995,456页)。如果节点是根节点,相同的频率集会输出两次。
注意,变量fix_kappa很少用到nhomo = 3,4或5。
fix_kappa = 1表示对所有分枝假定并估算一个共同的k,而fix_kappa = 0表示对每个分枝估算一个k。
nhomo = 2 适用K80,F84和HKY85模型(Yang 1994b;Yang 和 Yoder 1999)对树中的每个分枝估算一个转换/颠换比率(k)。
getSE = 0,1或2。 是否计估算计算参数的标准差。这里只是借助于参数相关的log似然值的二次导数的转置矩阵,使用曲率算法等进行粗略的计算。二次导数通过不同的方法计算,并不总是可靠。即使近似值是可靠的,依赖于SE的测试也要慎重,依赖正态近似值来估算最大似然率的测试也一样。似然率测试总是应该优先的。当执行树搜索时,选项不可用并选择getSE = 0。
RateAncestor = 0或1。通常使用0。值1是用来使程序进行两个额外的分析。如果你不需要这些结果可以忽略它。首先基于跨位点的变化率模型(如gamma,RateAncestor = 1)使程序采用经验贝叶斯步骤(Yang 和Wang 1995)沿序列来计算单独位点的比率(输出在文件rate中)。
第二,RateAncestor = 1使程序执行经验贝叶斯方法重构祖先序列(Yang等人 1995a;Koshi和Goldstein 1996;Yang 2006,199-124页)。祖先状态的重构采用著名的parsimony(Fitch 1971;Hartigan 1973)(通过PAML中的pamp程序执行)。它也可以用似然/经验贝叶斯法来完成。两个方法常产生相似的结果,但是基于似然法的重构有两个优势:它使用枝长信息和字符(核酸)间的相对替代率并且对重构的祖先状态提供一个后验概率形式的不确定性的尺度。
rst文件中的输出结果按位点排列。你可以使用变量verbose来控制输出的数量。如果verbose = 0,只有每个节点上每个位点最佳的核酸序列(具有最高后验概率的一条。)被列出。若verbose = 1(如果1不行试试2),边缘重构的后验概率分布都会被列出。如果模型对所有位点只假定一个比率,联合的和边缘的祖先重构都将被计算。如果模型在位点中假定变化的比率(如gamma模型),只有边缘重构被计算。
边缘和联合重构。边缘重构认为最好的重构方法应该是将字符分配给一个单独的内部节点并且这个字符拥有最高的后验概率(例如Yang等,1995a,4;或Yang ,2006,4.15)。边缘重构算法在PAML中可以同时在针对所有位点的恒定比率模型和位点比率的gamma模型下运行。联合重构认为最好的重构是所有的祖先节点在同一时间并且拥有最高的后验概率的重构(一个位点的字符集分配给所有的内部节点)(Yang,1995a,2;或Yang。2006,4.16)。PAML中的联合重构算法基于Pupko等人(2000)的工作,它能得到每个位点上的最佳重构和它的后验概率。基于这个模型的工作对全部位点只有一个比率(在分割模型下工作)。边缘和联合方法的使用虽略有不同,但预期产生的结果是一样的;也就是说,针对一个位点的联合重构法中的组成字符也最有可能是边缘重构中最佳的字符。当发生两个或多个重构序列具有相似的后验概率时,可能会有些冲突。
祖先序列重构的一个好用途就是综合推断祖先蛋白并在实验室中测量它们的生化性质(Pauling和Zuckerkandl 1963; Chang和Donoghue 2000;Thornton 2004)。将重构的祖先序列作为真实的观察数据来进行进一步分析也很常见。你应该抗拒这些不可抗拒的诱惑并使用全似然率方法,如果它们可用的话(如 Yang 2002)。参见Yang(2006)4.4.4章里对祖先重构中系统偏差的讨论。
对于蛋白编码DNA序列的核酸碱基分析(baseml)(序列文件中使用选项GC),程序也会计算祖先氨基酸的后验概率。这个分析中,枝长度和其它参数采用核酸替代模型估算,但是重构的核酸三联体作为一个密码子来对待以推断最可能的氨基酸编码。终止密码子的后验概率非常小并重置为0以便计算氨基酸的后验概率。使用这个选项,你需要在控制文件baseml.ctl中添加控制变量icode。这在上面没有给出。这个变量icode可以取值0,1,…11,以对应PAML中12中包含的遗传密码(参见控制文件codeml.ctl来定义不同的遗传密码)。核酸替代模型与密码子替代模型非常相近可以按如下指定。在数据文件第一行的末尾添加一个选项GC并选择model = 4(HKY85)和Mgene = 4。这样,模型对三个密码子位置假定不同的替代率,不同的碱基频率和不同的转换/颠换比率(kappa)。源自核酸替代的祖先重构与基于密码子的重构非常类似。
Small_Diff是一个非常小的数用在派生物的不同近似值中。
cleandata = 1表示移除全部序列中包含模糊的字符(不确定的核酸如N,?,W,R,Y等与四种核酸不同的其它类型)或比对空位的位点。cleandata = 0(默认的)则使用这些位点。
method:这个变量控制基于一个无分子钟模型的的估算枝长的迭代算法。method = 0执行PAML中的老算法,它更新所有参数,同时也包括枝长。method = 1指定PAML中的一个新算法,它一个接一个的更新枝长。method = 1在含有分子钟的模型下不能执行(clock = 1,2,3)。
icode:这用来指定蛋白编码DNA序列的祖先重构所使用的密码子。它将对带有基于密码子分析的祖先重构结果的比较。例如Goldman和Yang(1994)的F3×4密码子模型与对密码子的三个位置采用了不同替代速率和碱基频次的HKY85核酸模型非常相似。后者通过在序列数据文件中使用选项GC,model = 4和Mgene = 4来执行。要使用选项icode,你必须选择RateAncestor = 1
fix_blength:它将告诉如果树有枝长时程序该如何做。如果你想忽略枝长,那么使用0。如果你想程序从随机起始点开始,那么使用-1。如果你想使用枝长作为最大似然迭代的初始值,那么使用1。尽量避免使用PAUP中简约分析得来的枝长,它们对于整条序列(而不是每个位点)来说是个变化的数值并且是非常差劲的初始值。如果你想用给定的树文件中的枝长(而不是用ML来估算),那么使用2。这种情况下,你需要确定使用这些枝长是明智的。例如,如果数据文件中的两个序列是不同的,但是树中连接两个序列的两个枝长都为0,这个数据和树将有矛盾,程序将崩溃。
Output:输出不说也基本能懂。统计性的描述都会列出。观察位点模式和它们的频率连同通常模式的比例也会列出。每个物种的核酸比例(和多基因数据中每条基因)会被计算并列出。lmax = ln(lmax)是log似然率的上限,并可能与基于替代模型的最佳(或真实)树的似然率进行比较用以测试模型对数据的最佳拟合度(Goldman 1993;Yang等 1995b)。如果你不知道它表示什么你可以忽略它。输出中还包含成对序列的距离,一个叫做2base.t的单独文件中也包含它。这是一个下对角线矩阵,可以被Felesenstein的PHYLIP程序包中的NEIGHBOR程序读取。对模型JC69,K80,F81,F84使用合适的距离公式,而对于更复杂的模型使用TN93公式。Baseml主要是一个最大似然率程序,输出距离矩阵只是为了方便,对接下来的似然率计算没有什么用处。
getSE = 1时,S.E.s作为大样本方差的平方根来计算,并在参数估算下面精确地列出来。这行是0则表示错误,可能由于算法的分歧或枝长为0导致。
通常参数的S.Es用来衡量估算的可靠性。例如(k-1)/SE(k),当用K80估算出k,可以同正态分布比较来看K80和JC69是否有真正的不同。这个测试可以通过似然率测试来来更可靠的比较两个模型得出的log似然值。需要强调的是估算枝长的S.Es不能被曲解为是对树拓扑结构的可靠性的评估(1994c)。如果树文件中超过一棵树,程序baseml和codeml将采用RELL方法(Kishino和Hasegawa 1989)计算自举大小,也可以采用对多重比较带有校正的Shimodaira和Hasegawa(1999)的方法。自举重采样对合理的数据划分做出解释(在序列文件中使用选项G)。
5 basemlg
basemlg使用同baseml相同的控制文件baseml.ctl。由于不允许使用树搜索和假定的分子钟,因此要选择runmode = 0和clock = 0。basemlg可用的模型有JC69,F81,K80,F84和HKY85,并且对位点的比率总是假定一个gamma分布。变量ncatG,given_rho,rho,nhomo在这里无效。因为迭代过程中计算S.E.s,所以参数估计的S.E.s总是会输出,因此getSE变量也没有用。
由于basemlg需要密集计算,因此对于数据分析推荐baseml中的离散gamma模型。如果你选择使用basemlg,不需要首先运行baseml,然后在运行basemlg。这可以让baseml将初始值收集到一个名为in.basemlg的文件中,来供basemlg使用。注意,basemlg执行的只是baseml中模型的一个子集。
6 codeml(codonml和aaml)
密码子替代模型
这里有一系列密码子替代模型。详细的讨论参见Yang和Bielawski(2000),Yang(2002)和Yang(2006:第8章)。
核酸模型通常使用Goldman和Yang(1994)模型的简化版本并制定密码子i到密码子j的替代速率为

如果两个密码子在一个以上的位置不同。
同义颠换
同义转换
非同义颠换
非同义转换
(Yang等,1998)。密码子j的均衡频率(πj)可以被认为是一个自由参数,但也可以根据密码子三个位置上的核酸频率计算得到(控制变量 CodonFreq )。这个模型下,ω = dN/dS,也就是非同义/同义替代速率。对应这个碱基模型,需要在控制文件codeml.ctl中设定model = 0,NSites = 0。
ω比率用于度量蛋白的正选择作用。简而言之,ω值 < 1,= 1,> 1表示负的纯净选择,中性进化和正选择。然而所有位点的平均比率和所有的种系几乎从不 > 1,因为正选择不可能在漫长的时间中作用于所有的位点。因此,真正要探究的只是一些种系和一些位点所受的正选择影响。
分枝模型允许系统发育树分枝中使用多个ω比率,它用于检测个别种系承受的正选择作用(Yang 1998;Yang和Nielsen 1998)。它们用变量model指定。model = 1对应自由率模型,它假定每个分枝都有一个独立的ω比率。这个模型的参数非常丰富,不推荐使用。model = 2允许你有几个ω比率。你必须指定有多少个比率和树文件中用分枝标签来标定哪个分枝对应哪个比率。参见第四章“树文件格式”中的“枝或节点标签”。溶菌酶例子的数据在文件夹examples/lysozyme/里。查看说明文档。
表2. 位点模型的参数
| 模型 | NSsites | #p | 参数 |
| M0 (one ratio) | 0 | 1 | ω |
| M1a (neutral) | 1 | 2 | p0 (p1 = 1 – p0), ω0 < 1,ω1 = 1 |
| M2a (selection) | 2 | 4 | p0, p1 (p2 = 1 – p0 –p1), ω0 < 1,ω1 = 1,ω2 > 1 |
| M3 (discrete) | 3 | 5 | p0, p1 (p2 = 1 – p0 – p1) ω0,ω1,ω2 |
| M7 (beta) | 7 | 2 | p, q |
| M8 (beta&ω) | 8 | 4 | p0 (p1 = 1 – p0), p, q,ωs > 1 |
注意 #p是ω分布中自由参数的数量。括号中的参数不是自由的也不能被计算:例如,在M1a中,p1 = 1 – p0 它就不是自由参数。在比对M1a和M2a及M7和M8的似然率测试中,df = 2。位点模型通过使用NSsites指定。
位点模型允许位点中使用多个ω比率(蛋白中就是密码子和氨基酸)(Nielsen和Yang 1998;Yang等,2000b)。一些这样的模型通过设定codeml中的变量NSsites来运行(并且model = 0)。你可以通过为NSsites指定几个值来一次运行几个NSsites模型。例如,NSsites = 0 1 2 7 8在一次运行中对相同的数据计算5个模型。位点模型可用于真实数据的分析和计算机模拟研究的评估(Anisimova等,2001;Anisimova等,2002;Anisimova等,2003;Wong等,2004)。两对模型在进行正选择的两个似然率测试时似乎非常有用。第一对比对M1a(接近中性)和M2a(正选择),第二对比对M7(beta)和M8(beta和ω)。M1a(接近中性)和M2a(正选择)对模型M1(中性)和M2(选择)(Nielsen和Yang 1998)稍作了修改。两组测试中,要使用df = 2。M1a-M2a的比对似乎比M7-M8的比对更稳定。参见下面的表格。旧模型M1和M2设定ω0 = 1和ω1 = 1,而且不能对0 < ω < 1之间的位点进行计算。Wong等人(2004)和Yang等人(2005)的工作中描述的新模型M1a和M2a在设定ω1=1时可以估算出数据中的0 < ω0 < 1。版本3.14以后用于识别正选择位点的BEB程序也可以执行了。
第三组测试比对零假设的M8a(beta和ωs = 1)和M8(Swanson等,2003; Wong等,2004)。M8a用NSsite = 8,fix_omega = 1,omega = 1指定。零分布是50:50的由0和x1^2的混合体(self和Liang 1987)。5%的临界值为2.71,1%的是5.41(相对x1^2,5%的为3.84,1%的为6.63)。注意,50:50的xj^2和xk^2的混合体的p值只是两个分布的p值的平均数。在M8a-M8的比对中,我们从x1^2得到p值并且用它的一半来获得混合分布的p值。你也可以使用x1^2(Wong等,2004)。
我们建议M0-M3比对作为对位点中变量ω的测试而不是用于正选择测试。然而,对所有位点只用一个ω的模型在每一个功能蛋白中可能都会有错误,所以没有测验的点。
朴素经验贝叶斯(NEB)(Nielsen和Yang 1998;Yang等,2000b)与贝叶斯经验贝叶斯(BEB)(Yang等,2005)可用来计算位点类型的后验概率,如果似然率测试显著也可以用于识别正选择位点。NEB使用MLEs参数(如比例和ω速率)但不能计算它们的抽样误差,而BEB可以用先验贝叶斯处理抽样误差。BEB只能在M2a和M8下运行。我们建议你忽略NEB的输出,仅使用BEB的结果。
BEB输出结果格式如下:
Prob(w>1) mean w
135 K 0.983* 4.615 +- 1.329
解释:4.615是w后验分布的近似均值,1.329是w后验分布的方差的平方根。如果后验概率大于95%程序将输出*,如果大于99%则输出**。
Suzuki和Gojobori (1999)检测正选择位点的方法。使用的术语中,SG99方法检测每个位点的ω比率是否大于1或小于1。使用简约发重构祖先序列,那么对于每个位点,计算同义和非同义的差异数目以及同义和非同义的位点数。并测试位点上的ω与1有显著不同。祖先序列重构中的错误被忽略。Suzuki写了一个叫做AdaptSite的程序来执行这个测试。这只是一个直观的测试,缺乏说服力。
Codeml中,这种测试仅作为codeml或baseml中使用ML来重构祖先序列时的副产品来执行。使用RateAncestor = 1。与codeml相对的baseml的选择以及每个程序中替代模型的选择仅影响祖先序列的重构。接下来的步骤也一样,参照Suzuki和Gojobri(1999)。Codeml中我们使用M0(NSsites = 0和model = 0)。如果你愿意,你可以尝试其它模型,如NSsites = 2或8。典型的祖先重构模型几乎没什么差异。对于baseml,你需要在序列数据文件的第一行使用“GC”来指明序列是编码蛋白的。在控制文件中使用icode(= 0 用于普通密码子;= 1 用于脊椎动物线粒体密码子)来指定遗传密码子,如在codeml中。“多基因”模型与M0很接近:model = 4 Mgene = 4(参见Yang 1996a和“用于分段模型的结合分析”一节)
枝-位点模型允许蛋白中的位点和树中的枝的ω都可以变化并检测个别种系(称为前景枝)中影响很少位点的正选择。最初Yang和Nielsen执行模型A(model = 2 NSsites = 2)和模型B(model = 2 NSsites = 3)。模拟中(Zhang 2004)的测试并不顺利,因此构建了两个测试对模型A做了改动(表3)(Yang等,2005;Zhang等,2005)。测试2也是正选择的枝-位点测试,推荐使用这个测试。这个比较是将修改后的模型A与相对应的ω2设为1的空模型(fix_omega =1,omega = 1)对照的。零分布是由0和x1^2按50:50混合而成的,5%的临界值为2.71,%1的临界值为5.41。我们推荐替换x1^2(用临界值3.84和5.99)来防止模型假设的违例。
类似的都要对修改后的枝-位点模型A(不是模型B)执行针对位点类型计算后验概率的NEB和BEB方法。你需要结合BEB来使用模型A并忽略NEB的输出。
表3. 枝-位点模型的参数
| 位点类型 | 旧模型A (np = 3) | 新模型A (np = 4) | |||
| 比例 | 背景 | 前景 | 背景 | 前景 | |
| 0 | p0 | ω0 = 0 | ω0 = 0 | 0 <ω0 < 1 | 0 <ω0 < 1 |
| 1 | p1 | ω1 = 1 | ω1 = 1 | ω1 = 1 | ω1 = 1 |
| 2a | (1 – p0 – p1)p0/(p0 +p1) | ω0 = 0 | ω2>=1 | 0 <ω0 < 1 | ω2 >=1 |
| 2b | (1 – p0 – p1)p1/(p0 +p1) | ω1 = 1 | ω2>=1 | ω1 = 1 | ω2 >=1 |
注意 枝位点模型A用model = 2 Nssites = 2指定。枝位点测试的正选择是个可选的模型。空模型设定ω2 = 1。似然率测试 df = 1(见文本)。
进化枝模型通过model = 3 Nssites = 2指定,而进化枝模型D通过model = 3 Nssites = 3指定并用ncatG来指定位点类型的数量(Bielawski和Yang 2004;也可以参见Forsberg 和Christiansen, 2003)进化枝模型C做了些类似枝位点模型A所做的一些变化。新的模型C用 0 <ω0 < 1替换了ω0 = 0,并在ω分布中设置了5个参数:p0,p1,ω0,ω2和ω3。新的模型C可以同新的M1a(两个参数,d.f. ≈ 3)(接近中性)比较。
表4. 进化枝模型中的参数 (新旧对比)
| 旧模型C (np = 4) | 新模型C (np = 5) | ||||
| 位点类型 | 比例 | 进化枝1 | 进化枝2 | 进化枝1 | 进化枝2 |
| 0 | p0 | ω0 = 0 | ω0 = 0 | 0 <ω0 < 1 | 0 <ω0 < 1 |
| 1 | p1 | ω1 = 1 | ω1 = 1 | ω1 = 1 | ω1 = 1 |
| 2 | p2 = 1 – p0 –p1 | ω2 | ω3 | ω2 | ω3 |
进化枝模型D可以使用ncatG = 3 或 2。当指定枝位点模型A或B及进化枝模型C,并且模型中的类别数量确定时选项ncatG的变化是被忽略的。
BEB程序可以对模型C执行但不能用于模型D。你需要使用模型C与BEB程序结合。忽略NEB的输出。
模型C和模型D做了一个扩展允许针对两个以上的进化枝或分支类型。分支类型通过树文件中的标签指定。如果你有四个分支类型(标签#0,#1,#2,#3),进化枝模型如下。这里的ω2,ω3,ω4,ω5是(0,∞)范围中最佳的独立参数。
进化枝模型C下的BEB计算开销非常大(每个额外的分枝类型要增加10翻的计算量),因此只能用于分枝类型少的模型。
表5. 含两个以上分枝类型的进化枝模型C或D的参数
| 位点类型 | 比例 | 进化枝1 | 进化枝2 | 进化枝3 | 进化枝4 |
| 0 | p0 | 0 <ω0 < 1 | 0 <ω0 < 1 | 0 <ω0 < 1 | 0 <ω0 < 1 |
| 1 | p1 | ω1 | ω1 | ω1 | ω1 |
| 2 | p2 = 1 – p0 – p1 | ω2 | ω3 | ω4 | ω5 |
进化枝模型C中,ω1是固定的,而在进化枝模型D中,ω1是作为自由参数估算的。
突变-选择模型(Yang和Nielsen,2008)使用下面的控制变量执行。
CodonFreq = 7 * 0:1/61 each, 1:F1X4, 2:F3X4, 3:codon table
* 4:F1x4MG, 5:F3x4MG, 6:FMutSel0, 7:FMutSel
estFreq = 1
CodonFreq = 6 指定FMutSel0,7指定FmutSel。如果estFreq = 1,frequency/fitness参数通过ML从数据中估算,如果estFreq = 0,则用数据中的观察频率计算。模型的详细内容参见Yang和Nielsen (2008)。examples/mtCDNAape/文件夹中的说明文档说明了如何重复文献中表1的结果。
氨基酸替代模型
我对经验和机械的氨基酸替代模型做了区分(Yang 等,1998; 2006:第2章)。Codeml中的经验模型包括Dayhoff (Dayhoff 等1978),JTT (Jones 等1992), WAG (Whelan和Goldman 2001), mtMam (Yang 等1998), mtREV (Adachi和Hasegawa 1996a)等。它们从真实数据中按普通时间可逆模型估算替代率参数。
机械模型是考虑到氨基酸间的突变距离构成的,突变距离通过编码密码子的位置决定,对自然选择的突变的过滤是在蛋白水平上操作的(Yang等 1998)。Aaml程序实现了很少的这类模型,通过变量aaDist指定。
控制文件
由于基于密码子的分析和基于氨基酸的分析使用不同的模型,一些控制变量也就有了不同的意义,针对密码子和氨基酸序列使用不同的控制文件是个不错的主意。codeml默认的控制文件是codeml.ctl,如下:
seqfile = stewart.aa * sequence data file name
outfile = mlc * main result file name
treefile = stewart.trees * tree structure file name
noisy = 9 * 0,1,2,3,9: how much rubbish on the screen
verbose = 0 * 1: detailed output, 0: concise output
runmode = 0 * 0: user tree; 1: semi-automatic; 2: automatic
* 3: StepwiseAddition; (4,5):PerturbationNNI; -2: pairwise
seqtype = 2 * 1:codons; 2:AAs; 3:codons–>AAs
CodonFreq = 2 * 0:1/61 each, 1:F1X4, 2:F3X4, 3:codon table
* ndata = 10
clock = 0 * 0:no clock, 1:clock; 2:local clock; 3:TipDate
aaDist = 0 * 0:equal, +:geometric; -:linear, 1-6:G1974,Miyata,c,p,v,a
* 7:AAClasses
aaRatefile = wag.dat * only used for aa seqs with model=empirical(_F)
* dayhoff.dat, jones.dat, wag.dat, mtmam.dat, or your own
model = 2
* models for codons:
* 0:one, 1:b, 2:2 or more dN/dS ratios for branches
* models for AAs or codon-translated AAs:
* 0:poisson, 1:proportional,2:Empirical,3:Empirical+F
* 6:FromCodon, 8:REVaa_0, 9:REVaa(nr=189)
NSsites = 0 * 0:one w;1:neutral;2:selection; 3:discrete;4:freqs;
* 5:gamma;6:2gamma;7:beta;8:beta&w;9:betaγ
* 10:beta&gamma+1; 11:beta&normal>1; 12:0&2normal>1;
* 13:3normal>0
icode = 0 * 0:universal code; 1:mammalian mt; 2-11:see below
Mgene = 0 * 0:rates, 1:separate;
fix_kappa = 0 * 1: kappa fixed, 0: kappa to be estimated
kappa = 2 * initial or fixed kappa
fix_omega = 0 * 1: omega or omega_1 fixed, 0: estimate
omega = .4 * initial or fixed omega, for codons or codon-based AAs
fix_alpha = 1 * 0: estimate gamma shape parameter; 1: fix it at alpha
alpha = 0. * initial or fixed alpha, 0:infinity (constant rate)
Malpha = 0 * different alphas for genes
ncatG = 3 * # of categories in dG of NSsites models
fix_rho = 1 * 0: estimate rho; 1: fix it at rho
rho = 0. * initial or fixed rho, 0:no correlation
getSE = 0 * 0: don’t want them, 1: want S.E.s of estimates
RateAncestor = 0 * (0,1,2): rates (alpha>0) or ancestral states (1 or 2)
Small_Diff = .5e-6
* cleandata = 0 * remove sites with ambiguity data (1:yes, 0:no)?
* fix_blength = 0 * 0: ignore, -1: random, 1: initial, 2: fixed
method = 0 * 0: simultaneous; 1: one branch at a time
变量seqfile, outfile, treefile, noisy, Mgene, fix_alpha, alpha, Malpha, fix_rho, rho, clock, getSE, RateAncestor, Small_Diff, cleandata, ndata, fix_blength和method的使用与前面baseml.ctl中描述的一样。Seqtype在数据中指定序列的类型;seqtype = 1表示密码子序列(程序为codeml);2表示氨基酸序列(程序为aaml);3表示被翻译成蛋白的密码子序列。
密码子序列(seqtype = 1)
CodonFreq 在密码子替代模型中指定均衡的密码子频率。这些频率可以假设为相等的(每个标准遗传密码都是1/61,CodonFreq = 0),或从平均核酸频率计算得到(CodonFreq = 1),或来自三个密码子位置的平均核酸频率(CodonFreq = 2),又或使用自由参数(CodonFreq = 3)。这些密码子频率模型中参数的数值为0,3,9和60(通用密码子),分别对应CodonFreq = 0,1,2和3。
Clock 参见baseml控制文件。
aaDist 指定是否假定氨基酸距离是相等的(= 0)或使用Grantham矩阵(= 1)(Yang等,1998)。论文中用于分析的线粒体数据示例在软件包中的example/mtdna文件夹。aaDist = 7(AAClasses),可以使用密码子和氨基酸序列来运行,允许你有一些氨基酸替代类型并让程序估算它们的不同速率。Yang等,(1998)使用了这个模型。替代类型的数量和氨基酸对的变化属于那种类型是在叫做OmegaAA.dat的文件中指定的。你可以对“保守”和“变化”的氨基酸替代情况使用模型来配合不同的ω速率。examples/mtCDNA中的例子是对应这个模型的。查看文件夹内的说明文件。
Runmode = -2 执行ML来估算成对比较的蛋白编码序列中的ds 和dn(seqtype = 1)。程序将估算出的ds和dn结果输出到2ML.ds和2ML.dn。由于许多使用者似乎对种系中的dn/ds比率很感兴趣,那么他们可能也对通过计算两个比率得到的枝长来表示的树型的检验感兴趣,尽管这个分析有点特别。如果你的物种名不超过10个字符,你可以使用输出的距离矩阵作为Phylip程序的输入,这不需要任何改动。否则你需要编辑文件将名字裁短。对于氨基酸序列(seqtype = 2),选项runmode = -2使程序在替代模型下通过数值迭代来计算ML距离,要么在所有位点使用一个比率的模型下(alpha = 0),要么在位点的比率为gamma模型下(alpha ≠ 0)。在后者中,使用连续的gamma并且变量ncatG被忽略。(runmode = 0,使用离散gamma。)
Model指定枝模型(Yang 1998;Yang 和 Nielsen 1998)。Model = 0表示对所有分枝使用一个比率;1表示每个分枝一个比率(自由比率模型);2表示任意数目的比率(如2-ratios或3-ratios模型)。当model = 2时,你必须将树中的分枝用符号#或$进行分组。参见前面关于指定枝位点模型的章节。Model = 2,变量fix_omega设定最后的ω比率(ωk-1如果总计有k个比率)为文件中指定的omega的值。这个选项用于测试作为前景枝的比率是否显著于其它的值。参见examples/lysozyme文件夹来重复Yang(1998)中的结果。
NSsites 指定位点模型,NSsites = m相当于Yang(2000b)中的Mm模型。变量ncatG用于指定一些模型下ω分布中的类别数量。在Yang等(2000b)的工作中,3对应M3(离散),5对应M4(频率),10对应连续分布(M5:gamma,M6:2gamma,M7:beta,M8:beta&w,M9:beta&gamma,M10: beta&gamma+1, M11:beta&normal>1, and M12:0&2normal>1, M13:3normal > 0)。例如M8有11个位点类别(10个来自beta分布加上1个额外的用于正选择的类别,同时ωs≥1)。参见前面密码子替代模型章节中关于Paml3.14中M1和M2变化的部分。
你可以在一个分枝中运行几个NSsites模型。如下,在Yang等(2000b)中,使用默认的ncatG并使用
NSsites = 0 1 2 3 7 8
这些位点的后验概率以及这些位点预期的ω值列在了rst文件中,它可能用于识别正选择位点。
参见examples/hivNSsites/和examples/lysine/下使用位点模型分析的例子。
枝位点模型A(参见前面的密码子替代模型章节)通过使用变量model和NSsites指定。
Model A: model = 2, NSsites = 2, fix_omega = 0
对枝位点正选择测试的模型是可选的。空模型也是枝位点模型,但是设定了ω2 = 1,并指定
Model A1: model = 2, NSsites = 2, fix_omega = 1, omega = 1
这里有些值得注意的是对与两个旧的测试我们并不推荐。旧的枝位点模型B(Yang 和 Nielsen 2002)通过下面参数指定。
Model B: model = 2, NSsites = 3
旧测试B的空模型是含2种位点类型的NSsites模型3(离散):
model = 0, NSsites = 3, ncatG = 2
使用d.f. = 2。Yang和Nielesn(2002)的大规模的溶解酶数据集分析在examples/lysozyme文件夹中。
同样适用于Yang等(2005)中描述的枝位点测试1和Zhang等(2005)使用的修改后的枝位点模型A作为被择假设,而零假设是d.f. = 2的新位点模型M1a(接近中性)。当前景枝在宽松的选择约束或正选择压力时这个测试是显著的。建议使用测试2来替代它,测试2也称为正选择的枝位点测试。
Bielawski和Yang (2004)的进化枝模型C和D通过下面方式指定:
Model C: model = 3, NSsites = 2
Model D: model = 3, NSsites = 3 ncatG = 2
详细内容参见文献。类似的模型C也进行了修改并且BEB程序只能用于模型C。参见前面。
Icode 指定遗传密码。十一个遗传密码表通过使用icode = 0到10来应用,它们分别对应于GeneBank中的transl_table 1到11。0为通用密码子;1为哺乳动物线粒体密码子,3为霉菌线粒体;4为无脊椎线粒体;5为纤毛虫细胞核密码子;6为棘皮动物线粒体;7为euplotid线粒体;8为替代酵母细胞核;9为海鞘线粒体;10为赭纤虫细胞核。还有一种额外的密码子,叫做Yang氏规则密码子,通过icode = 11来指定。这套密码子中有16种氨基酸,密码子第一位和第二位的偏差为非同义替代,第三位的偏差为同义替代。也就是说所有的密码子都是四重简并。目前没有任何生物体使用这套密码子。
Mgene 同序列文件中的选项G结合使用,按表6中概括的来指定分割模型(Yang 和 Swanson2002)。文章中使用的溶菌酶数据包含在examples/文件夹中。分析将溶解酶中的氨基酸分为包埋的和暴露的两部分(“基因”),并在分析中对各部分的异质性做了解释。你可以阅读说明文件并重复表6中Yang和Swanson(2002)的结果。
表6. 密码子替代的分割模型的设置
| 序列文件 | 控制文件 | 跨越基因的参数 |
| 无G | Mgene = 0 | 每个都相等 |
| 选项 G | Mgene = 0 | (k,ω)和π相同,但cs不同。(成比例的枝长) |
| 选项 G | Mgene = 2 | (k,ω)相同,但πs和cs不同。 |
| 选项 G | Mgene = 3 | π相同但(k,ω)和cs不同 |
| 选项 G | Mgene = 4 | (k,ω),πs和cs都不同 |
| 选项 G | Mgene = 1 | 单独分析 |
Fix_alpha,alpha:对于密码子模型,fix_alpha和alpha为位点指定gamma比率模型,模型中密码子中针对位点变化的相关比率服从gamma分布,但是ω比率在全部位点中保持不变。这是对核酸和氨基酸gamma模型的一个懒惰的扩展。我不喜欢这个模型并且建议你用NSsites模型来替代它(使用NSsites变量来指定,并且设定fix_alpha = 1,alpha = 0)。如果你对这个程序同时指定NSsites和alpha,它将中止运行。
RateAncestor:参见baseml控制文件中的描述。
密码子序列(seqtype = 1)的输出:每个序列中的密码子频率被计算并列在遗传密码表中,以及它们在各个物种中的总和。每个表格包含六个或更少的物种。对于多基因数据(序列文件中指定了选项G),每个基因中的密码子频率也会被列出。密码子位置上的核酸分布也会列出。Nei和Gojobori (1986)的方法用来计算每个同义替代位点(ds)上发生了同义替代的数量以及每个非同义替代位点(dn)上发生了非同义替代的数量及它们的比值(dn/ds)。这些用于构建对似然性分析中枝长的最初估算,但是它们不能似然性估算自身。
祖先重构(RateAncestor)的结果在rst文件中。在计算位点中变量dn/ds比率的模型里(NSsites模型),位点类型的后验概率和正选择位点也都在rst文件中。
氨基酸序列(seqtype = 2)
Model指定氨基酸替代模型:0为泊松模型,它假设所有的氨基酸替代是均等的速度(Bishop 和 Friday 1987);1为比例模型,该模型中一个氨基酸改变的速度是与氨基酸的频率成比例的。Model = 2指定一类经验模型,先验的氨基酸替代速率矩阵通过aaRatefile文件指定。文件包含在软件包中用于Dayhoff(dayhoff.dat),JTT,WAG(wag.dat),mtMAM(mtmam.dat),mtREV24(mtREV24.dat)等的经验模型。
如果你想指定自己的替代速率矩阵,可以参看这些文件,里面注释了文件的结构。氨基酸替代模型的其它选择被忽略。简而言之,变量model,aaDist,CodonFreq,NSsites和icode用于指定密码子模型(seqtype = 1),而model,alpha和aaRatefile用于氨基酸序列。
Mgene 同序列文件中的选项G结合使用,按表6中概括的来指定分割模型(Yang 和 Swanson2002)。文章中使用的溶菌酶数据包含在examples/文件夹中。分析将溶解酶中的氨基酸分为包埋的和暴露的两部分(“基因”),并在分析中对各部分的异质性做了解释。你可以阅读说明文件并重复表6中Yang和Swanson(2002)的结果。(此处与上面完全相同,原文中如此,可能是作者编辑时出现了错误。)
表7. 氨基酸替代的分割模型的设置
| 序列文件 | 控制文件 | 跨越基因的参数 |
| 无G | Mgene = 0 | 每个都相等 |
| 选项G | Mgene = 0 | π相同,但cs不同。(成比例的枝长) |
| 选项G | Mgene = 2 | πs和cs都不同 |
| 选项G | Mgene = 1 | 单独分析 |
Runmode 同baseml.ctl中的工作方式一致。指定runmode = 2将使程序在成对比较中计算ML距离。你可以改变控制文件中的变量aaRatefile,model和alpha。
如果你进行成对的ML比较(runmode = -2)并且数据包含模糊字符或者对齐空位如果cleandata = 1,程序将从所有序列中移除这类的位点。这也称为“全部删除”。如果cleandata = 0,将成对的移除对齐空位和模糊字符。{{这似乎不是真的。如果runmode = -2,程序当前移除所有模糊的位点。需要检查。注意ziheng 31/08/04}}注意,在种系发生的多序列的似然性分析中,如果cleandata = 0,对齐空位被当做模糊字符对待,如果cleandata = 1,对齐空位和模糊字符都会被删除。注意移除对齐空位和将它们作为模糊字符对待都会降低序列分歧。数据中的模糊字符(cleandata = 0)使似然率计算变慢。
氨基酸序列(seqtype = 2)输出:输出文件不需解释,它与核算和密码子分析的结果非常相似。氨基酸替代的经验模型(通过dayhoff.dat,jones.dat,wag.dat,mtmam.dato或mtREV24.dat指定)在替代速率矩阵中不包括任何参数。当RateAncestor = 1时,祖先重构的结果在rst文件中。用位点变化速率模型计算位点替代速率的结果在rates文件中。
7 evolver
这个程序生成一个简单的菜单,如下:
(1) Get random UNROOTED trees?
(2) Get random ROOTED trees?
(3) List all UNROOTED trees into file trees?
(4) List all ROOTED trees into file trees?
(5) Simulate nucleotide data sets (use MCbase.dat)?
(6) Simulate codon data sets (use MCcodon.dat)?
(7) Simulate amino acid data sets (use MCaa.dat)?
(8) Calculate identical bi-partitions between trees?
(9) Calculate clade support values (read 2 treefiles)?
(0) Quit?
选项 1,2,3,4。这个程序可以用于生成一个随机树,无根或有根都可以,也可以有枝长或无枝长。当然,你只能用于少量的物种否则你的硬盘将被无用的树塞满。选项8用于从一个树文件中读取多个数并且计算两部分的距离,要么是第一个和剩余的树之间,要么是每对之间。
选项9(进化枝支持值)可用于概括自举和贝叶斯分析。它读入两个树文件。第一个树文件只能包含一棵树,如最大似然树。你在文件的前端需要有物种的数量和树的数量(只能是1棵树的)。第二个树文件可以包含树的集合,比如从1000次自举假样本中估算出的1000个最大似然树。这个选项会为树文件里的ML树中的每个进化枝计算自举支持值,也就是说,第二个树文件中的比例树包含第一个文件中树的进化枝或节点。第二个树文件中的第一行并不必须含有树和物种的数量。如果你运行MrBayes,你可以将最大似然树或最大经验树移动到第一个文件中,第二个树文件可以由MrBayes生成.t文件来替代,不需要任何改动。现在物种只是用树文件中的数字来表示。你可以选择这个选项9来运行evolver。程序将向你询问两个输入文件的名字。另一种方法是绕开这个简单的菜单,而在命令行中输入选项和两个文件的名字:evolver 9 <MasterTreeFile> <TreesFile>
选项 5,6,7(模拟)evolver程序模拟带有用户指定树拓扑结构和枝长的核酸,密码子和氨基酸序列。用户在控制文件中指定替代模型和参数。见下面。程序在一个文件中以PAML(输出mc.paml)或PAUP(mc.paup)格式生成多个数据集如果你选择PAUP格式,程序将寻找名为paupstart(程序将其拷贝到数据文件的前面),paupblock(程序将其拷贝到每个模拟数据集的末尾)和paupend(程序将其合并在文件的末尾)文件。
这可以是PAUP程序在一次运行中分析全部数据集。模拟的参数在三个文件中指定:MCbase.dat, MCcodon.dat和MCaa.dat分别对应核酸,密码子和氨基酸序列。运行默认的参数并在屏幕输出中查看。然后查看appropriate .dat 文件。作为例子,MCbase.dat文件在下面列出。注意文件的第一块作为evolver的输入,其它的为注释。树的长度是种系发生中沿着所有分枝的每位点替代数的期望值,计算为枝长的总和。当我用模拟来评估序列分歧的效率又要保持树型的固定时,引入了这个变量。Evolver将测量树来计算指定树长的枝长。如果你对树长使用-1,程序将使用树中给定的枝长而不再重新计算。还要注意碱基频率必须按固定的顺序。MCaa.dat和MCcodon.dat中的氨基酸与密码子频率也一样。
0 * 0: paml format (mc.paml); 1:paup format (mc.nex)
367891 * random number seed (odd number)
5 1000000 1 * <# seqs> <# nucleotide sites> <# replicates>
-1 * <tree length, use -1 if tree has absolute branch lengths>
(((A :0.1, B :0.2) :0.12, C :0.3) :0.123, D :0.4, E :0.5) ;
3 * model: 0:JC69, 1:K80, 2:F81, 3:F84, 4:HKY85, 5:T92, 6:TN93, 7:REV
5 * kappa or rate parameters in model
0 0 * <alpha> <#categories for discrete gamma>
0.1 0.2 0.3 0.4 * base frequencies
T C A G
Evolver的模拟选项(5,6,7)可以使用命令行格式避开简单的菜单。因此下面是一些运行evolver可能的方式。
evolver
evolver 5 MyMCbaseFile
evolver 6 MyMCcodonFile
evolver 7 MyMCaaFile
密码子替代模型使用选项6来假设系统发生树中所有分枝和基因上的所有位点都具有相同的ω比率。这就是所谓的M0模型(一比率)。在位点中使用带有ω变量的位点模型来模拟,或者在分枝中使用带有不同ω的分枝模型,又或在位点和分枝中使用变化的ω的枝位点模型,请阅读paml/Technical/Simulation/Codon/文件夹下的CodonSimulation.txt文件。
第一个变量控制使用的序列数据格式:0表示paml/phylip格式,1位点模式记数和2nexus格式。位点模式记数可以被baseml或codeml读入,如果你有带有很多位点的大规模数据(>106),这是很有用的。(参见前面的序列数据格式部分)
Evolver也可以对每个模拟数据使用带有随机枝长的随机树进行模拟。你必须改变源代码并重新编译。打开evolver.c然后找到函数Simulate中的fixtree = 1并将它改成0。重新编译并将程序命名为evolverRandomTree,如。
cc -o evolverRandomTree -O2 evolver.c tools.c –lm evolver 5 MCbaseRandomTree.dat
控制文件如MCbase.dat也需要改变。软件包中含有一个名字为MCbaseRandomTree.dat的例子文件。“树长”和树拓扑结构的一行被出生率λ,死亡率μ,样本比率ρ和树高所替代(从根到顶端的每个位点替代数的期望值)。树用带有物种样本的生-灭过程生成(Yang和Rannala 1997)。时钟是假设的。你必须改变源码来释放时钟。如果你对文件格式选择0(paml),随机树将输出在文件ancestral.txt中;它可以用Rod Page的TreeView查看。如果文件格式选择2(nexus),程序将把树输出在序列文件的树的一块内容中。
Evolver程序还有几个选项用来列出几个少量物种的所有的树,并从分枝进化,含有物种样本的生-灭过程模型中生成随机树(Yang和Rannala 1997)。它还有一个选项来计算树拓扑结构间的分歧距离。
Evolver中使用的蒙特卡罗模拟算法。你可以在“模型与分析”一节中读到更相信的内容。参见第九章中Yang(2006)。这有一些简短的注释。Evolver通过沿着树的“进化”序列模拟数据集。首先,靠模型或数据文件指定使用均衡的核酸,氨基酸或密码子频率对根生成一个序列(MCbase.dat,MCcodon.dat和MCaa.dat)。每个位点按照枝长、替代模型的参数等沿着树枝进化。当序列中的位点按照相同的过程进化,转移概率矩阵对每个枝的所有位点只计算一次。对所谓的位点-分类模型(如gamma,NSsites密码子模型),不同位点可能拥有不同的转移概率矩阵。枝起始处的字符和结尾处的字符是从通过源字符得到的转移概率所指定的多项式分布中抽样得来的。通过模拟生成祖先节点的序列并输出在ancestral.txt文件中。
一些人想在根处指定序列而不是让程序随机生成序列。这可以通过在RootSeq.txt中放入一条序列来实现。这个序列不能包含模糊字符或空位或终止密码子。在几乎所有的模拟中,确定根序列是完全错误的,因此你需要避免犯这种错误。如果你想通过模拟来反映你的特殊基因,你可能要用一个模型从那个基因来估算参数并使用参数估计来模拟数据集。
8 yn00
程序yn00执行Yang和Nielsen(2000)的方法来估算两个序列间的同义和非同义替代速率(ds和dn)。Nei和Gojobori(1986)的方法也包含在内。程序中的这个专门方法用来解释颠换/转换比的偏差和密码子使用偏差,它是假设密码子频率模型为F3×4时的ML法的近似值,用来解释颠换/转换比率速率。我们推荐你尽可能使用ML法(控制文件中runmode= -2,CodonFreq = 2),即使对成对比较的序列也是。
seqfile = abglobin.nuc * sequence data file name
outfile = yn * main result file
verbose = 0 * 1: detailed output (list sequences), 0: concise output
icode = 0 * 0:universal code; 1:mammalian mt; 2-10:see below
weighting = 0 * weighting pathways between codons (0/1)?
commonf3x4 = 0 * use one set of codon freqs for all pairs (0/1)?
上面展示了一个yn00的控制文件,指定了序列文件的名称(seqfile),输出文件的名称(outfile)和遗传密码子(icode)。序列中包含对齐空位和模糊核酸的位点将从全部的序列中移除。变量weighting决定当在密码子之间计算时使用相等的权重或不等的权重。两种方法对相异的序列是不同的,不等的权重将计算的较慢。从数据文件中对所有序列估算颠换/转换比速率并用于随后的成对比较。Commonf3×4指定密码子频率是从数据文件中的每对序列间估算还是所有的序列中估算。除了主要的结果文件,程序还声称三个距离矩阵:同义替代率的2YN.ds,非同义替代率的2YN.dn,结合密码子比率的2YN.t(t是作为每密码子核酸替代率的数目来估算的)。这些都是下对角线矩阵并且可被一些诸如Felesenstein 开发的PHYLIP中的NEIGHBOR之类的程序直接读取。
原文来自:http://blog.csdn.net/winglyx/article/details/6731787
1F
在Linux系统下编译软件时,rm *.o命令执行不成功,请问是什么原因