

基因表达分析里面,RNA-seq是现在转录组研究常用的技术了,但是通过二代测序获得数据后,在正式分析前我们通常需要做两件事情:其一是reads的饱和度分析,另一个是RNA-seq测序的数据与mRNA真实表达水平之间的一致性。今天生物信息学QQ群里面(ID:235461986,欢迎从事生物信息学同行加入群讨论)刚好有人问到这个问题,所以这里就整理一下我的观点。另外,本来大家还讨论了关于基因组组装、宏基因组测序、以及非编码RNA方面方面的深度测序问题,但是个人感觉,信息量不够大,这次就不把这些方面的内容混着一起放上来了。待后续讨论比较详细或者相关内容比较丰富的时候我再做个整理放上来。
下面回到reads的饱和度分析方面。我以自己一个实际研究的课题来简单介绍一下我的方法。我做的是细菌的转录组。测序完之后,数据处理的基本流程
1)对低质量的reads进行过滤
我的研究中采用的是非链特异性测序,single-end的reads,reads读长81bp。一般测序的reads中,每条read后面的一些碱基可能会测得比较差(从测序的reads质量报告图上可以观察得到)。所以做转录组研究中可以把后面那些质量差的截掉。
设定的过滤条件:
- read前20bp无N(这一条可以考虑不要)
- 整条read中质量值低于20的碱基不超过20%
- 整条read中质量值低于13的碱基不超过10%
- 整条read的碱基平均值不小于20
对于不满足条件的reads,采取的办法就是不断去掉最后一个碱基;去掉之后分析剩下前N-1bp的碱基分析,利用上面的条件进行筛选,如还是不满足继续截断,然后对剩下N-2bp分析,由此下去,当最后长度小于50bp的时候,如果还不满足条件的话,直接把该条reads丢掉,然后分析下一条reads。
以上筛选标准是我们实验室常用的筛选标准,大家可以进入生物信息学QQ群(235461986)发表自己的看法,或者P直接在PLoB网站评论留下自己的筛选方法。这种方法适用于转录组,做基因组拼接组装的就不要截短reads,不符合条件的reads的直接删掉,原因嘛,自己可以想一想噢,呵呵。
2)reads做mapping,以及筛选
reads做mapping的软件很多了,例如bwa等等,PLoB网站上面也有很多关于bwa等软件的安装、使用教程,大家可以自己去搜一下。这里就不赘述了。
得到sam文件之后,选取那些uniq mapping的reads。
补充一个方面:其实在1)中,设定最短reads长度cutoff的时候,其实就有一个长度设多少合适的问题。我只能说,我处理数据的时候设定的是50bp。有人会问可以更短一些么,当然可以,这样至少会有一个方面的问题:当reads太短的话,最后这条reads可能会落到多个地方,不是uniq mapping的最后会被删掉。另外对于single end的数据,可能会错误mapping。当然如果是pair-end的话我想会好很多。毕竟人家SOLID测出35bp也可以做转录组。
3)饱和度评估
得到sam文件后其实我们可以计算出每一条reads落在哪里,基因间区或者基因区(基因区的话在哪个基因上也可以列出来)。假如我们得到3,200,000条uniq mapping的reads。我们可以采用梯度为10,000,分别为100,000, 200,000, 300,000, 。。。3,100,000, 3,200,000,依次随即抽样,看抽出来的这些reads分别检测到多少基因。然后把reads数和检测的基因数画一个曲线,看看这条曲线是否饱和。若,当reads数到一定程度检测到的基因几乎不增加或者很少增加,则测序包和,否者就是测序量不够没有达到饱和。
下面给与一个实际的例子供大家参考,例子中屏蔽了相关四个样本信息,请大家理解。
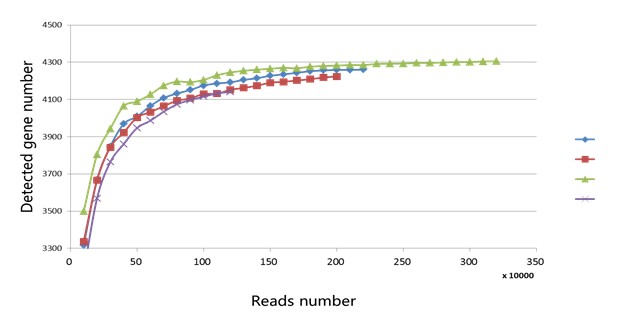
其实我们可以参考别人论文中测序数据量评估一下自己在做转录组的时候需要测多少数据量。但是注意一点就是,rRNA的影响会比较大。据我了解从cell中提出来的total RNA中90%是rRNA。rRNA是对我们做转录组分析没有意义的,所以我们需要去掉,但是不一定去得很好,我感觉能去掉50%就不错了。对于一些特殊的细菌,现在的试剂盒在去16sRNA和23sRNA去得不好。我就遇到这样问题,直接导致测序没饱和,所有样品重测了一次。更多内容欢迎大家到群里面讨论。
补充一:
如何计算reads覆盖的基因数?
其实这地方我认为有一个比较好的办法就:首先将该样品所有的reads都mapping到基因组上去,然后根据他们mapping的位置可以知道每个reads落在哪里,基因间区或者基因区(哪个基因都可以详细计算出来)。在抽样的过程中,就随机选取一定数量的reads,看看有多少reads落在基因上,然后用hash表计算一共有多少个基因。
1F
请问,具体如何操作啊?